
Abstract
Two samples of acid-hydrolyzed nanocellulose and two samples of TEMPO-oxidized nanocellulose were separately prepared from cotton liner pulp and microcrystalline cellulose, and dispersed in water. Sodium alginate that was extracted from brown seaweed was dissolved in the nanocellulose suspensions and wet spun in a calcium chloride bath to form four kinds of alginate/nanocellulose composite fibers. The structures and properties of the obtained nanocellulose and composite fibers were investigated and compared. The results showed that all of the nanocellulose samples exhibited a needle shape with slightly different sizes. The incorporation of nanocellulose increased the opacity of the spinning dopes but improved the mechanical properties of the alginate fibers. The optimum addition amount for all of the nanocelluloses was 5% (based on the weight of sodium alginate). The TEMPO-oxidized nanocellulose produced from cotton liner pulp had the greatest influence on the strength of the fibers. All the composite fibers had an irregular cross-section with dense and uniform structure, which indicated the good compatibility between nanocellulose and alginate. In addition, the introduction of nanocellulose slightly improved the thermal stability of the alginate fibers.
Download PDF
Full Article
Comparison of Acid-hydrolyzed and TEMPO-oxidized Nanocellulose for Reinforcing Alginate Fibers
Xiao-Jun Shen,a,b,c# Pan-Li Huang,c# Jing-Huan Chen,d,* Yu-Ying Wu,c Qiu-Yun Liu,e and Run-Cang Sun c,*
Two samples of acid-hydrolyzed nanocellulose and two samples of TEMPO-oxidized nanocellulose were separately prepared from cotton liner pulp and microcrystalline cellulose, and dispersed in water. Sodium alginate that was extracted from brown seaweed was dissolved in the nanocellulose suspensions and wet spun in a calcium chloride bath to form four kinds of alginate/nanocellulose composite fibers. The structures and properties of the obtained nanocellulose and composite fibers were investigated and compared. The results showed that all of the nanocellulose samples exhibited a needle shape with slightly different sizes. The incorporation of nanocellulose increased the opacity of the spinning dopes but improved the mechanical properties of the alginate fibers. The optimum addition amount for all of the nanocelluloses was 5% (based on the weight of sodium alginate). The TEMPO-oxidized nanocellulose produced from cotton liner pulp had the greatest influence on the strength of the fibers. All the composite fibers had an irregular cross-section with dense and uniform structure, which indicated the good compatibility between nanocellulose and alginate. In addition, the introduction of nanocellulose slightly improved the thermal stability of the alginate fibers.
Keywords: Sodium alginate; Acid-hydrolyzed nanocellulose; TEMPO-oxidized nanocellulose; Enhancement; Alginate fiber
Contact information: a: Beijing National Laboratory for Molecular Sciences, Key Laboratory of Colloid and Interface and Thermodynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China; b: University of Chinese Academy of Sciences, Beijing 100049, China; c: Beijing Key Laboratory of Lignocellulosic Chemistry, Beijing Forestry University, Beijing 100083, China; d: National Engineering Laboratory of Pulping and Papermaking, China National Pulp and Paper Research Institute, Beijing 100102, China; e: The BioComposites Centre, University of Wales, Bangor LL57 2UW, UK LL57 2UW; *Corresponding authors: jinghuanchen@126.com; rcsun3@bjfu.edu.cn
# These authors contributed equally to this work
INTRODUCTION
Recently, polysaccharides with their valuable properties, including non-toxicity, biocompatibility, biodegradability, and water solubility, were shown to be ideal sources of polymeric materials (Lin et al. 2012b; Sibaja et al. 2015). Alginate, which can be isolated from natural brown seaweeds, is a linear, unbranched, anionic polysaccharide, consisting of two C5 epimer units, namely β-D-mannuronic acid (M) and α-L-guluronic acid (G) (Pawar and Edgar 2012). Typically, the monomer units are present in three different polymer segments: GG, MM, and alternating MG residues. The carboxyl functional groups of G-blocks could form ionic bonds with divalent cations, such as Ca2+, Ba2+, and Cu2+, leading to the transformation of an alginate solution into a stable gel (Grant et al. 1973). Thus, the gels prepared from alginates with a high content of G-blocks (especially in GG segment) have a considerably higher strength than those prepared from alginates rich in M-blocks. Alginate fibers could be produced in a calcium chloride bath via a wet spinning method, and have been extensively used in medical dressings due to their remarkable bioactivity, hygroscopicity, oxygen permeability, and non-toxicity (Qin 2008; Zhao et al. 2015). However, its further applications are limited due to their poor mechanical properties.
Cellulose is the most abundant natural polymer on earth and can be extracted from a variety of plants. During the last two decades, nanocellulose materials have attracted significant interest because of their unique advantages, including large specific surface area, high strength and stiffness, low density, and other nanoscale properties (Domingues et al. 2014). Some specific characteristics, such as surface charge and dimensions, may vary depending on the preparation methods and type of cellulose raw materials (Jonoobi et al. 2015). Numerous methods have been reported for nanocellulose production. Nanocrystalline cellulose consists of rod-like crystalline particles of 10 to 30 nm in width and several hundred nanometers in length, and usually produced by strong acid hydrolysis (Habibi 2014). Sulfuric acid hydrolysis is a typical method used to isolate nanocellulose with good dispersibility in water. This method is generally based on the dissociation of microscale celluloses in concentrated sulfuric acid (mass fraction of ∼60%) for a short period of time (e.g., an hour) at mild temperatures (e.g., ∼40 oC), followed by several post-treatment steps, such as centrifugation, dialysis, and ultrasonication. This method can produce cellulose nanocrystals with a length of ∼100 nm and a characteristic cross-sectional dimension of ∼20 nm (Zhou et al. 2011; Gu et al. 2013; Habibi 2014; Huan et al. 2016). Due to the sulfuric etherification reaction, hydroxyl groups on the surface of cellulose crystals are randomly replaced by sulfate ester groups, where the resulting system can form a stable colloidal suspension (Miao et al. 2017). Different from sulfuric acid hydrolyzed nanocellulose, TEMPO-oxidized nanocellulose have long flexible fiber networks and consists of alternating crystalline and amorphous cellulose domains. TEMPO-oxidized nanocellulose are mainly produced by 2,2,6,6-tetramethylpiperidine-1-loxy (TEMPO)-mediated oxidation or mechanically induced deconstructing strategies. The CNFs obtained by TEMPO-mediated oxidation are more uniform and can be well dispersed in an aqueous phase (Habibi et al. 2006). It is worth noting that the TEMPO oxidation can selectively oxidize hydroxyl groups at the C6 position on the cellulose surface and create carboxylate groups, where the dispersed nanocellulose particles can also form a colloidal suspension in an aqueous environment. The resulting cellulose nanofibers usually have smaller cross-sectional dimensions (2 to 8 nm) and longer length. In short, the surface of nanocellulose prepared from a sulfuric acid hydrolysis method contains negatively charged sulfate groups (Dong et al. 1996), while nanocellulose prepared from TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl)-mediated oxidation contain significant amounts of C6 carboxylate groups (Habibi 2014). These groups are advantageous to the dispersion and stability of nanocellulose suspensions (Dong et al. 1998; Huq et al. 2012). Additionally, various kinds of nanocellulose have been used to improve the mechanical properties and thermal stability of polymer matrices (Siró et al. 2010; Lin et al. 2012b), such as cellulose acetate (Lu and Drzal 2010), chitosan (Nordqvist et al. 2007; Hassan et al. 2011), amylopectin (Plackett et al. 2010), and polyvinyl alcohol (Lu et al. 2008; Cai et al. 2016).
The similar chemical structure of nanocellulose and alginate could provide good compatibility for their composite materials (Yang et al. 2000). In a previous work, acid-hydrolyzed nanocellulose isolated from cotton with an average length of 130 nm and an average width of 20.4 nm was introduced into the calcium alginate fibers (Ureña-Benavides et al. 2010). The mechanical testing of the fiber showed a 38% increase in tenacity and a 123% increase in tensile modulus when 10 wt.% nanocellulose was loaded, indicating that nanocellulose is a potential enhancer to reinforce alginate fibers. The effect of nanocelluloses prepared from different methods and materials on the properties of alginate fibers needs to be further investigated.
In this study, four kinds of nanocelluloses were prepared from cotton liner pulp (CP) and microcrystalline cellulose (MCC) by acid hydrolyzation and TEMPO oxidization methods and used to enhance the alginate fibers. Normally, nanocellulose suspensions are added dropwise into the spinning dopes of sodium alginate under vigorous stirring, but this does not guarantee the uniform distribution of the nanocellulose due to the high viscosity of the dopes. For this reason, a new preparation method was adopted, namely nanocellulose specimens were first dispersed in water, and sodium alginate powders were then dissolved in the nanocellulose suspension. The obtained spinning dopes were then wet spun into a calcium chloride bath to form composite fibers. The morphology of the prepared nanocellulose was characterized by atomic force microscopy (AFM). The chemical structure, mechanical, and thermal properties of alginate fibers were investigated and compared by gel permeation chromatography (GPC), proton nuclear magnetic resonance (1H NMR), Fourier transform infrared spectroscopy (FT-IR), X-ray diffraction (XRD), scanning electron microscopy (SEM), tensile testing, and thermogravimetric (TG) analysis. The purpose of this study was to give a comprehensive understanding of the effect of nanocelluloses’ structure on the properties of alginate fibers.
EXPERIMENTAL
Materials and Preparation Methods
Brown seaweed, collected from the Shandong province, China, was selected as the raw material to extract sodium alginate. It was exhaustively washed with tap water, air-dried, and cut into pieces before use. The CP with α-cellulose content of more than 95% was provided by Hubei Chemical Fiber Co. Ltd. (Hubei Province, China). The MCC was obtained from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). All other chemicals, including TEMPO, formaldehyde (HCHO), ethanol (CH3CH2OH), sulfuric acid (H2SO4), hydrochloric acid (HCl), sodium bromide (NaBr), sodium hypochlorite (NaClO), anhydrous calcium chloride (CaCl2), and anhydrous sodium carbonate (NaCO3), were of analytical grade and used without further purification (Beijing Chemical Company, Beijing, China).
Extraction of sodium alginate
The extraction of sodium alginate from brown seaweed was performed according to previous literature (Gomez et al. 2009) with minor modifications. Firstly, 5 g of air-dried seaweed samples were soaked in 300 mL of 1 wt.% formaldehyde solution at room temperature (25 °C) for 4 h to fix the pigments on the surface. Then, the seaweeds were washed with distilled water and treated with 300 mL of 4 wt.% Na2CO3 at 46 °C for 3 h under constant stirring (300 rpm). After reaction, the sodium alginate solution was collected from the reaction mixture by filtering, neutralized (pH 6 to 7) with HCl. 10 wt.% CaCl2, and then added dropwise into the filtrate to precipitate alginate in calcium salt form. The obtained calcium alginate was washed thoroughly with distilled water and then acidified with 5 wt.% HCl for 1 h. Next, the gel materials of alginic acid were obtained by filtering and washed several times with distilled water. The alginic acid was then dispersed in distilled water and converted into sodium alginate by neutralization (pH 6 to 7) with 1 wt.% NaOH. Finally, the sodium alginate was precipitated and purified by ethanol, and collected for further use after freeze-drying.
Preparation of the nanocellulose
The acid-hydrolyzed nanocellulose was prepared from MCC and CP according to the methods described previously (Bondeson et al. 2006; Ben Azouz et al. 2012) with slight modifications. Briefly, 5.0 g cotton liner pulp was treated with 65 mL of 64 wt.% H2SO4 at 45 °C for 45 min under mechanical stirring, while 5.0 g MCC was treated with 50 mL of 63 wt.% H2SO4 at 50 °C for 35 min. After acid hydrolysis, the suspension was diluted with 10-fold cold deionized water to quench the reaction. The particles were precipitated for several hours and separated by centrifugation at 10000 rpm for 10 min. The resulting precipitates were dialyzed against deionized water for several days to remove the low molecular weight carbohydrates formed during hydrolysis, excess acid, and other water-soluble impurities until the pH value of the precipitates was around 7. The obtained nanocellulose was diluted and dispersed completely by ultrasonic equipment (JY99-IIDN, NanJing Immanuel Instrument Equipment Co., Ltd., Nanjing, China), and then labeled as NCa (using MCC as raw material) and NCb (using CP as raw material).
The TEMPO-oxidized nanocellulose were prepared from MCC and CP according to a previous method (Isogai et al. 2011). First, 5.0 g cellulose (MCC or cotton linter pulp) was dispersed in 500 mL deionized water by ultrasonic treatment. Then, TEMPO (78 mg, 0.5 mmol) and NaBr (0.515 g, 5 mmol) were added into the suspension and stirred magnetically at room temperature. The reaction was initiated by adding NaClO (8.0 g, 18 mmol) as a primary oxidant. The pH value of the suspension was maintained constantly at 10.5 by adding 0.5 M NaOH solution. At the end of the reaction, the oxidized cellulose was filtered and washed with deionized water. Subsequently, the oxidized cellulose was put into the deionized water and was placed in an ultrasonic cell disruptor (Scientz-IID, Ningbo, China) with 500 W power for 15 min at 0 oC to obtain oxidized nanocellulose, which was labeled as NCc (using MCC as raw material) and NCd (using CP as raw material), separately.
Preparation of alginate/nanocellulose composite fibers
Firstly, 0, 1, 5, 10, 15, 20, and 25% of the nanocelluloses (based on the weight of sodium alginate) were separately dispersed in water by ultrasonication. Then, 0.5 g of sodium alginate powder was directly added into the nanocellulose suspension under vigorous stirring until homogeneous dispersion was formed. The final concentration of alginate in the dope was 4.0 wt.%. After being vacuum degassed, the spinning dope was transferred to a syringe with the needle of 0.21-mm diameter and extruded into a 5 wt.% CaCl2 coagulating bath. The formed fibers were washed several times with distilled water to remove the excessive salts, and then collected by a spool and air-dried. The obtained fibers were labeled as F0, Fa-x, Fb-x, Fc-x, and Fd-x, in which the subscript a, b, c, and d corresponded to the added nanocellulose NCa, NCb NCc, and NCd, x represents the amount of nanocellulose, and F0 represents the alginate fiber without nanocellulose.
Characterization methods
Characterization of sodium alginate
The molecular weights of sodium alginate were determined by gel-permeation chromatography (GPC) on a PL aquagel-OH 50 column (300 mm by 7.7 mm, Polymer Laboratories Ltd., Redwood City, CA, USA), with a differential refractive index detector (RID), calibrated with PL pullulan polysaccharide standards (Mw of 738, 12,200, 100,000, and 16,00,000, Polymer Laboratories Ltd., Redwood City, CA, USA). The eluent was 0.02 mol/L NaCl in 0.005 M sodium phosphate buffer (pH 7.5) at a flow rate of 0.5 mL/min and kept at 30 °C. Sodium alginate was dissolved in 0.005 M sodium phosphate buffer (pH 7.5) with 0.02 mol/L NaCl at a concentration of 0.1%.
The 1H NMR spectra was recorded on a Bruker AVIII 400 MHz (Karlsruhe, Germany) spectrometer at 25 °C. Sodium alginate (200 mg) was dissolved in 50 mL of distilled water, acidified to pH 3.0 with HCl and heated at 90 °C for 1 h to reduce the viscosity of the solution. The solution was adjusted to neutral by the addition of NaOH and freeze-dried. The freeze-dried sodium alginate (10 mg) was then dissolved in 1 mL of D2O for G, M, GG, MM, and GM blocks determination according to a previous publication (Grasdalen et al. 1979).
Characterization of nanocellulose
The morphology and size distribution of the nanocellulose were investigated by atomic force microscopy (AFM) on a Nanoscope IIIa multimode scanning probe microscope (Digital Instruments Inc., Billerica, USA). The initial samples were diluted to 0.02 wt.% and sonicated for 10 min. A drop of suspension was then deposited onto the mica substrate and air-dried. The images were collected by using a tapping mode regime with silicon cantilevers (Bruker Co. Ltd., Karlsruhe, Germany) and scanning from 1 μm to 10 μm in air under room temperature. The images were only flattened without any other modification. Besides, the particle size of nanocellulose was determined by soft of Particle size distribution calculation 1.1, and the number of single nanocellulose was 100.
Characterization of spinning dopes and fibers
The dissolution states of the obtained spinning dopes were observed by an optical microscope (Nikon eclipse E200, Tokyo, Japan). The viscosity of the spinning dopes was measured on a Brookfield digital viscometer DV-II + PRO (Brookfield Instruments, Phoenix, USA) using a S34 rotor with a speed of 0.3 rpm at room temperature. The surface and cross-section morphologies of the fiber samples were collected by a scanning electron microscope (SEM) (S-3400N, Hitachi, Tokyo, Japan) at an acceleration voltage of 5 kV. Before the observation, the fibers were fractured in liquid nitrogen, stuck onto a sample platform, and sputtered with gold-palladium in a sputter coater (E-1010, Hitachi, Tokyo, Japan).
The FT-IR spectra of the fibers was scanned on a Thermo Scientific Nicolet iN10 FT-IR Microscope (Thermo Nicolet Corporation, Waltham, USA) equipped with a MCT detector cooled by liquid nitrogen in the reflection mode. The spectra were recorded in the range from 4000 cm-1 to 670 cm-1 with 64 sans at a 4 cm-1 resolution.
The crystalline structure of the nanocellulose and fibers were determined on a XRD-7000 X-ray diffractometer (Shimadzu, Tokyo, Japan) with Cu Kα radiation source (λ = 0.154 nm) at 40 kV and 30 mA. The X-ray patterns were acquired from 5° to 40° (2θ) with a scanning speed of 2°/min and a step interval of 0.02°. The crystallinity index (CrI) was calculated based on the height of the peak corresponding to (002) lattice plane (I002) and the minimum between 110 and 002 lattice planes (Iam) following equation, Eq. 1 (Segal et al. 1959):
![]() (1)
(1)
Mechanical properties, including tensile strength and elongation at break, of the fibers were measured according to ASTM D2256-80 (1980) with a UTM6503 universal tensile machine (Shenzhen Suns Technology Stock Co., Ltd., Shenzhen, China) equipped with a 100 N load cell. All of the samples were placed in 65% relative humidity (RH) and 20 °C for 24 h previously, and tested with 20-mm gauge length at a strain speed of 2.0 mm/min. The tensile strength values (δ) were calculated from the ratio of fracture force (F) and linear density (l) as follows,
![]() (2)
(2)
where F (cN) is the average force and l (dtex) is the linear density (represents the weight of fibers 10000 m in length). The linear density was estimated by the accurate weight of fibers 90 cm in length.
The thermal behavior of the fibers was studied by thermogravimetric analysis (TGA) using a DTG-60 simultaneous thermal analyzer (Shimadzu, Tokyo, Japan) under nitrogen atmosphere. Approximately 3 mg to 6 mg samples were loaded in an alumina crucible and heated from 40 °C to 900 °C at the rate of 10 °C/min.
RESULTS AND DISCUSSION
Chemical Structure of the Extracted Sodium Alginate
The physical properties of alginate, including molecular weight, M/G ratio, as well as the monomer sequence of M and G across the macromolecular chain, were usually dependent on the species of raw materials and the extraction methods. In this study, the weight-average molecular weight (Mw) of the obtained sodium alginate was 1.51×106 g/mol, which was much larger than that of sodium alginate prepared from other methods (HCl-Na2CO3 method, CaCl2-HCl-Na2CO3 method, etc.) as published previously (Chee et al. 2011; Fertah et al. 2014; Sellimi et al. 2015). Meanwhile, it was found that high molecular weight alginate-based materials exhibit excellent mechanical strength, thermal stability, and antioxidant properties (Draget et al. 1994; Şen 2011).
Table 1. Molecular Weight and Chemical Composition of Sodium Alginate Isolated from Seaweed in Laboratory
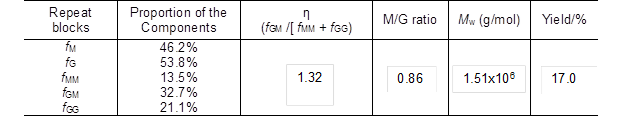
Quantification of the sodium alginate sample by 1H NMR can provide explicit structural information. The chemical composition and monomer sequence (M and G) of sodium alginate are related to the species of algae used. It has been found that alginate extracted from Tunisia brown seaweed C. barbata is composed of 37% mannuronic acid and 63% guluronic acid (Sellimi et al. 2015). The ratios of monomer M and G (M/G) of alginates extracted from different brown seaweed sources are 0.31 for S. muticum from England, 0.52 for S. fluitans from Cuba, and 0.94 for S. turbinarioides Grunow from Madagascar (Fenoradosoa et al. 2010). In this study, the M/G value of the extracted alginate was 0.86 (Table 1), which was higher than the values reported in previous studies (below 0.7) (Fenoradosoa et al. 2010; Sellimi et al. 2015). In addition, the distribution of M and G blocks could be illustrated by an η parameter (η = fGM / [fMM + fGG]) according to a previous study (Grasdalen et al. 1979). Alginate with a low η value (0 ≤ η < 1) corresponded to a main sequence distribution of MM or GG-sequences, while a high η value (1 < η ≤ 2) was related to a major sequence of alternate GM-blocks. The formation of gels depends mainly on the auto-cooperatively formed junctions between chain regions enriched in GG-sequences. As listed in Table 1, the η parameter of the sodium alginate in this paper was 1.32, which suggested that the sequence distribution of the obtained sodium alginate was mainly the alternate block (GM) type.
Morphology of the Obtained Nanocellulose
Normally, the size of the nanocellulose depends on the DP of raw materials, the preparation methods, and conditions. The AFM images and length distributions of the prepared nanocellulose samples are shown in Fig. 1. The little aggregates observed from the AFM images were probably formed on the mica during the drying procedure. All the nanocellulose samples exhibited a needle-shape morphology with a diameter of approximately 40 nm and length of 50 nm to 440 nm. The length distribution and average length of the four nanocellulose samples were slightly different. The nanocelluloses prepared from CP (the average length of NCb and NCd were 154 and 198 nm, respectively) were a little longer than those prepared from MCC (the average length of NCa and NCc were 120 and 136 nm, respectively). For the same raw material, the length of TEMPO-oxidized nanocellulose was slightly higher than that of the acid-hydrolyzed nanocellulose, which was in agreement with a previous report (Jonoobi et al. 2015). The TEMPO-oxidized nanocellulose produced (the average length of NCc and NCd were 154 and 198 nm, respectively) in this study was shorter than that prepared from the same method in other studies (about 1000 nm) (Montanari et al. 2005; Miao et al. 2016), which was possibly ascribed to the long oxidation time used. Differences in the size of nanocellulose would affect the performance of the alginate/nanocellulose composite fibers.
Properties of the Spinning Dopes
The complete dissolution of sodium alginate in the suspension of nanocellulose was the key step in the preparation of spinning dopes. Figure 2 shows the optical microscope images and digital photographs of the spinning dopes with different amounts of nanocellulose. With the increase of nanocellulose content, the color of the dopes was gradually changed from transparent to white. Meanwhile, the viscosity of the spinning dopes increased remarkably (Table 2). Figure 2 shows that the sodium alginate powders were fully dissolved in water (Fig. 2A), and that no obvious insoluble substances were observed in dopes when the addition amount of nanocellulose was less than 5% (Figs. 2B through 2C). However, when the addition amount was higher than 5%, a small amount of insoluble substances appeared in the solutions (Figs. 2D through 2F). These substances were likely to be the self-aggregations of nanocellulose due to the high concentration, and would affect the morphology, mechanical strength, and thermal stability of the prepared alginate fibers.
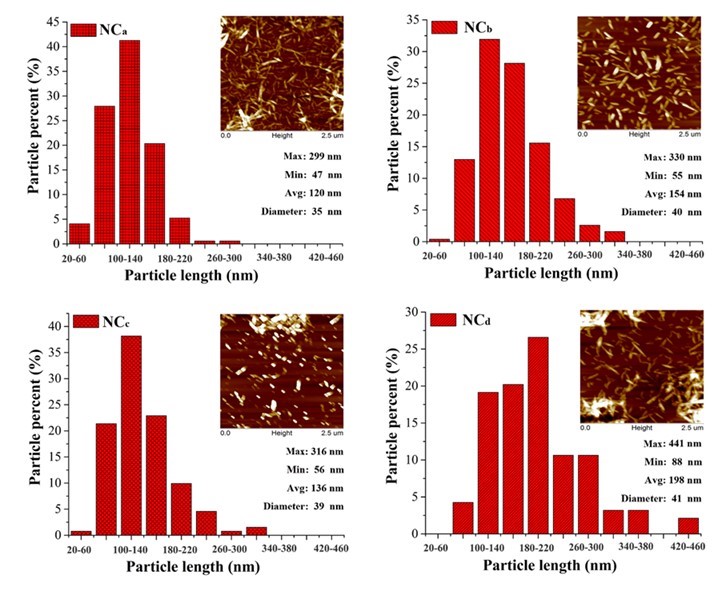
Fig. 1. Distribution histograms of the lengths of the nanocellulose and its AFM images (Min., Max., and Avg. present the minimum, maximum, and average length of the nanocellulose, respectively)
Table 2. Viscosity of Spinning Dopes with Different Amount of Nanocellulose
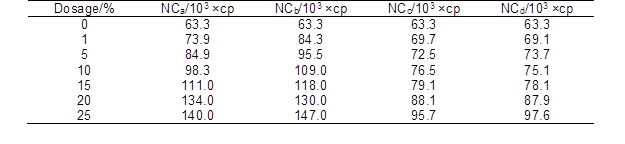

Fig. 2. Digital photographs and optical microscope images of spinning dope with different amounts of NCb: (a) 0%, (b) 1%, (c) 5%, (d) 10%, (e) 15%, and (f) 25%
Chemical Structure of the Alginate/Nanocellulose Fibers
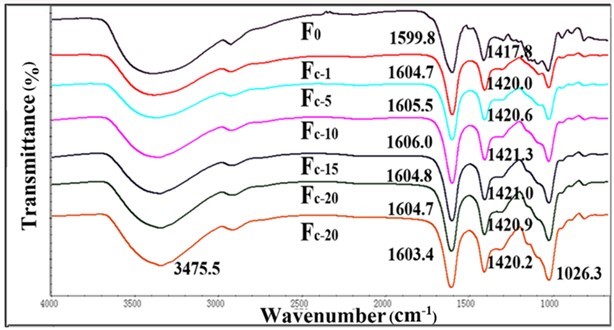
Fig. 3. FTIR spectra for Fc and pure alginate fiber (F0)
Fourier transform infrared spectroscopy is often used to investigate the chemical structure of materials. The FT-IR spectra of the pure alginate fiber and composite fibers are shown in Figs. 3, 4, S1, and S2. The spectrum of the pure alginate fiber (F0) showed a wide absorption band at 3400 cm−1, which was attributed to –OH groups, while the bands at 1599.8 cm-1 and 1417.8 cm-1 originated from the stretching vibration of –COO. Simultaneously, the signals at 1020 cm-1 corresponded to –C-O-C-. As the nanocellulose content increased, the peaks of -C=O (1599.8 cm-1) and -COO- (1417.8 cm-1) shifted toward a higher wavelength, which indicated that either the sulfonic group or carboxylic group in nanocellulose could link with adjacent alginate molecules to form a crosslinking network.
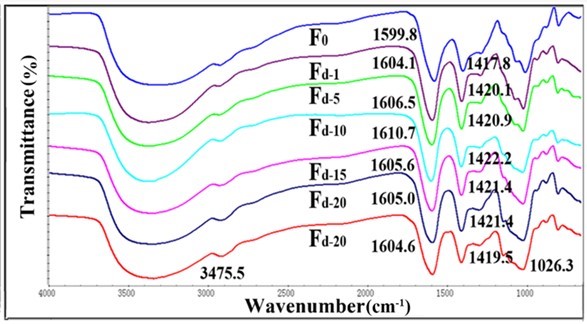
Fig. 4. FTIR spectra for Fd and pure alginate fiber (F0)
The shifts of the fibers containing TEMPO-oxidized nanocellulose were more evident than that of fibers containing acid-hydrolyzed nanocellulose. This might be attributed to the better ionic interactions and molecular compatibility of carboxylic nanocellulose as compared with sulfonic nanocellulose (Lin et al. 2012a). The FT-IR analysis of the fibers confirmed the intermolecular and ionic interactions between alginate and nanocellulose components. However, when the content of nanocellulose was higher than 10%, the –C=O absorption peak was no longer shifted to a higher wavelength. This might be because the cross-linked structure in alginate fibers was disrupted by the excessive amount of nanocellulose.
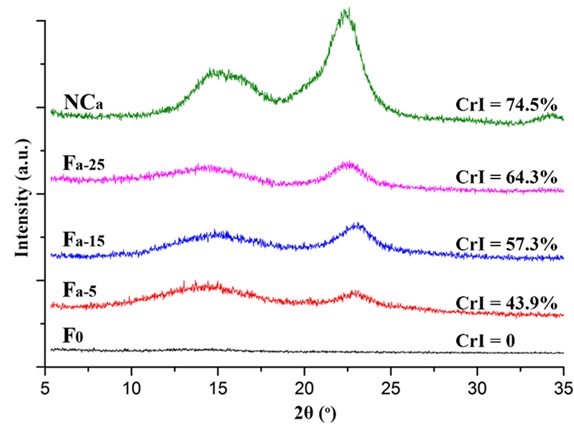
Fig. 5. XRD patterns of the NCa and Fa
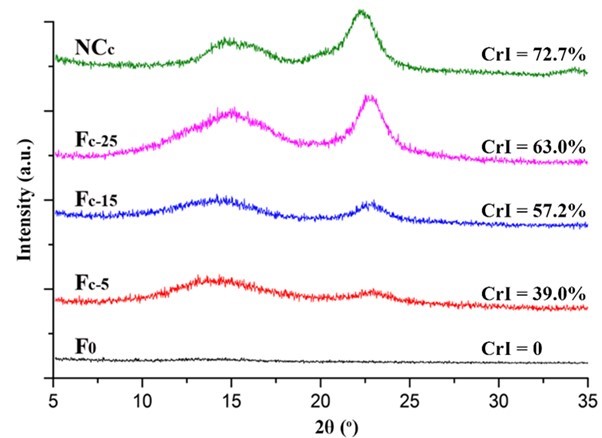
Fig. 6. XRD patterns of the NCc and Fc
The crystalline structure of the nanocellulose and the composite alginate fibers was characterized by XRD. As shown in Figs. 5, 6, S3, and S4, all of the prepared nanocellulose exhibited a characteristic pattern of cellulose I. The CrI of the acid-hydrolyzed nanocellulose (NCa and NCb) was slightly higher than that of the TEMPO-oxidized nanocellulose (NCc and NCd). The pure alginate fibers (F0) displayed an amorphous structure. The CrI of the composite fibers gradually increased with the increment of nanocellulose content.
Morphological Structure of the Alginate/Nanocellulose Fibers

Fig. 7. Cross-section morphology of the alginate fibers with various nanocelluloses
The cross-section and surface morphology of the pure alginate and composite fibers are displayed in Figs. 7, 8, S5, and S6. All of the fibers showed an irregular cross-section with an approximate 40 μm diameter. The density and homogeneity of the fibers were not influenced by the addition of nanocellulose. The pure alginate fiber (F0) had a rough surface with stripes along the fiber direction, which is formed due to the shrinkage of fiber during the drying process (Watthanaphanit et al. 2010). From the magnified photos in Fig. 8, a wave-like surface was observed in the alginate fiber. With the increment of nanocellulose content, the stripes on the surfaces of the composite fibers became deeper and irregular. This may have been because the redundant nanocellulose was hardly dispersed homogeneously in the water and spinning dopes and aggregated in the fibers.

Fig. 8. Surface morphology of alginate/nanocellulose fibers
Mechanical Properties of the Fibers
The mechanical properties of the composite fibers were influenced by the incorporation of nanocellulose. The tensile strength and elongation at break of pure alginate fiber (F0) was 1.12 cN/dtex and 16.05%, respectively (Figs. 9 and 10). With the increment of nanocellulose content, the tensile strength of the fibers was first increased and then gradually decreased. The highest strength values were obtained when the nanocellulose content was 5%. The reason for the decrease in fiber strength might be that the overdosed nanocellulose were easily agglomerated, which weakened the combination within the fibers and even disrupted the “egg-box” assembly in alginate (Ureña-Benavides et al. 2010). In general, the enhancement of TEMPO-oxidized nanocellulose was superior to that of the acid-hydrolyzed nanocellulose, and CP was the more appropriate material to prepare the nanocellulose for enhancing the alginate fiber as compared to MCC. The highest tensile strength value (1.64 cN/dtex) of the composite fiber was achieved when 5% NCd was added. The improvement of tensile strength might be ascribed to the good interfacial interaction and compatibility between the nanocellulose and alginate matrix. Figure 10 shows the elongation at break of the composite fibers. The addition of nanocellulose, especially NCa and NCc, markedly increased the elongation at break of the fibers. In addition, it should be noted that all fibers in this paper were spun in a low speed wet spinning manner without stretching, which did not fully exploit the potential enhancement of nanocellulose.
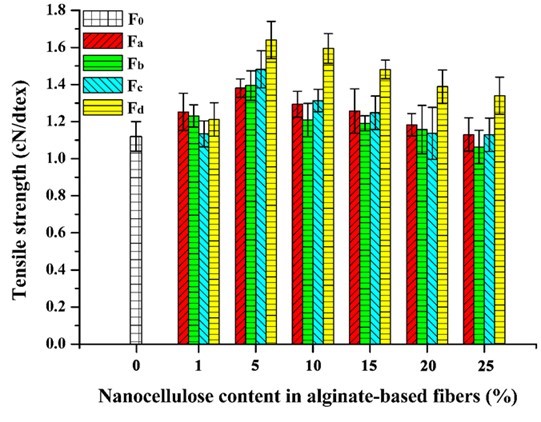
Fig. 9. Tensile strength of the alginate/nanocellulose fibers
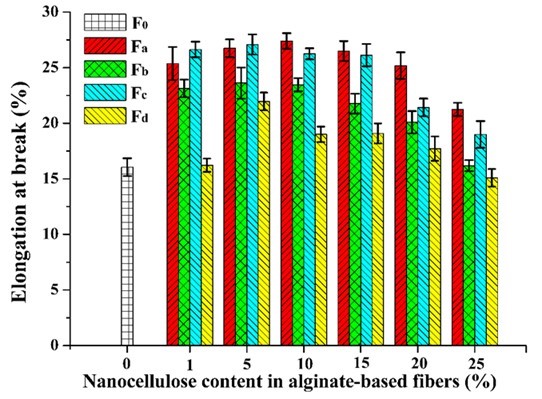
Fig. 10. Elongation at break of the alginate/nanocellulose fibers
Thermal Properties of the Fibers
The TG and DTG analyses were performed to evaluate the thermal stability of the fibers. As shown in Figs. 11 and 12, the first weight loss of all of the fibers in the temperature range from 40 °C to 200 °C corresponded to the evaporation of absorbed water (Parikh and Madamwar 2006; Stojanovic et al. 2012). The second temperature drop stage at 200 °C to 325 °C was due to the thermal destruction of the glycosidic bonds of alginate. The weight loss in the temperature range of 325 °C to 400 °C was ascribed to the destruction of the glycosidic bonds of nanocellulose. The small weight loss between 400 °C and 570 °C was contributed to the oxidation of intermediate carbonaceous char material formed in the third temperature drop stage (Parikh and Madamwar 2006). When the oxidation process was complete, a mass loss was apparent in the temperature range of 700 °C to 900 °C, which is associated with the partial decomposition of calcium carbonate into calcium oxide and carbon dioxide (Soares et al. 2004; Parikh and Madamwar 2006; Li et al. 2011). The thermo-decomposition rate for alginate in the fibers decreased in the ranges of 40 °C to 200 °C and 200 °C to 325 °C with the increase of nanocellulose content. Meanwhile, the weight residues of the composite fibers were between 21% and 23.9%, which was higher than that of the alginate fiber (19%). However, the decomposition temperatures of all the composite fibers within the range of 700 °C to 900 °C shifted to lower values possibly due to the incorporation of nanocelluloses. In contrast, the thermal stability of the fibers containing TEMPO-oxidized nanocellulose was slightly better than that of the fibers containing acid-hydrolyzed nanocellulose when the nanocellulose content was fixed (Fig. S5). Therefore, the thermal property of the composite fibers was influenced by the type and amount of nanocellulose.
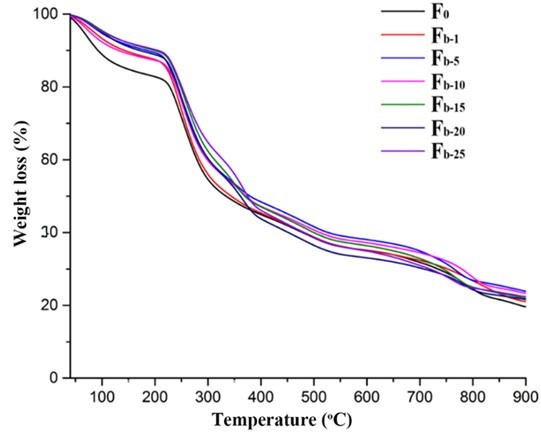
Fig. 11. TGA analysis of the alginate/nanocellulose fibers
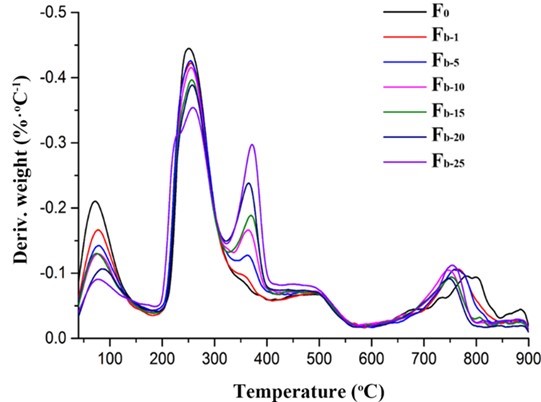
Fig. 12. DTG analysis of the alginate/nanocellulose fibers
CONCLUSIONS
- The morphology, structure, and performance of the obtained fibers revealed that the incorporation of nanocellulose increased the opacity and viscosity of the spinning dopes, but it did not influence the dissolution state of alginate.
- The obtained composite fibers had a dense irregular cross-section and stripe structure surface due to the compatibility of alginate and nanocellulose.
- The mechanical properties and thermal property of the fibers were influenced by the type and amount of the nanocellulose added.
- Approximately 5% of this nanocellulose was added, the tensile strength of the fibers increased 46%, and the fibers’ thermal stability was also improved.
- In general, the TEMPO-oxidized nanocellulose prepared from CP was the best choice to enhance alginate fibers.
ACKNOWLEDGMENTS
The authors are grateful for the support of National Natural Science Foundation of China for a key project (31430092), International Science & Technology Cooperation program of China (2015DFG31860), and Fundamental Research Funds for the Central Universities (2015ZCQ-CL-O2).
REFERENCES CITED
ASTM D2256-80 (1980). “Standard test method for breaking strength and elongation of pressure-sensitive tape,” ASTM International, West Conshohocken, PA.
Ben Azouz, K., Ramires, E. C., Van den Fonteyne, W., El Kissi, N., and Dufresne, A. (2012). “Simple method for the melt extrusion of a cellulose nanocrystal reinforced hydrophobic polymer,” ACS Macro Letters 1(1), 236-240. DOI: 10.1021/mz2001737
Bondeson, D., Mathew, A., and Oksman, K. (2006). “Optimization of the isolation of nanocrystals from microcrystalline cellulose by acid hydrolysis,” Cellulose 13(2), 171-180. DOI: 10.1007/s10570-006-9061-4
Cai, J., Chen, J., Zhang, Q., Lei, M., He, J., Xiao, A., Ma, C., Li, S., and Xiong, H. (2016). “Well-aligned cellulose nanofiber-reinforced polyvinyl alcohol composite film: Mechanical and optical properties,” Carbohydrate Polymers 140, 238-245. DOI: 10.1016/j.carbpol.2015.12.039
Chee, S.-Y., Wong, P.-K., and Wong, C.-L. (2011). “Extraction and characterisation of alginate from brown seaweeds (Fucales, Phaeophyceae) collected from Port Dickson, Peninsular Malaysia,” Journal of Applied Phycology 23(2), 191-196. DOI: 10.1007/s10811-010-9533-7
Domingues, R. M. A., Gomes, M. E., and Reis, R. L. (2014). “The potential of cellulose nanocrystals in tissue engineering strategies,” Biomacromolecules 15(7), 2327-2346. DOI: 10.1021/bm500524s
Dong, X. M., Kimura, T., Revol, J.-F., and Gray, D. G. (1996). “Effects of ionic strength on the isotropic−chiral nematic phase transition of suspensions of cellulose crystallites,” Langmuir 12(8), 2076-2082. DOI: 10.1021/la950133b
Dong, X. M., Revol, J.-F., and Gray, D. G. (1998). “Effect of microcrystallite preparation conditions on the formation of colloid crystals of cellulose,” Cellulose 5(1), 19-32. DOI: 10.1023/A:1009260511939
Draget, K. I., Bræk, G. S., and Smidsrød, O. (1994). “Alginic acid gels: The effect of alginate chemical composition and molecular weight,” Carbohydrate Polymers 25(1), 31-38. DOI: 10.1016/0144-8617(94)90159-7
Fenoradosoa, T. A., Ali, G., Delattre, C., Laroche, C., Petit, E., Wadouachi, A., and Michaud, P. (2010). “Extraction and characterization of an alginate from the brown seaweed Sargassum turbinarioides Grunow,” Journal of Applied Phycology 22(2), 131-137. DOI: 10.1007/s10811-009-9432-y
Fertah, M., Belfkira, A., Dahmane, E. M., Taourirte, M., and Brouillette, F. (2014). “Extraction and characterization of sodium alginate from Moroccan Laminaria digitata brown seaweed,” Arabian Journal of Chemistry (In press). DOI: 10.1016/j.arabjc.2014.05.003
Gomez, C. G., Lambrecht, M. V. P., Lozano, J. E., Rinaudo, M., and Villar, M. A. (2009). “Influence of the extraction–purification conditions on final properties of alginates obtained from brown algae (Macrocystis pyrifera),” International Journal of Biological Macromolecules 44(4), 365-371. DOI: 10.1016/j.ijbiomac.2009.02.005
Grant, G. T., Morris, E. R., Rees, D. A., Smith, P. J. C., and Thom, D. (1973). “Biological interactions between polysaccharides and divalent cations: The egg-box model,” FEBS Letters 32(1), 195-198. DOI: 10.1016/0014-5793(73)80770-7
Grasdalen, H., Larsen, B., and Smidsrød, O. (1979). “A p.m.r. study of the composition and sequence of uronate residues in alginates,” Carbohydrate Research 68(1), 23-31. DOI: 10.1016/S0008-6215(00)84051-3
Gu, J., Catchmark, J. M., Kaiser, E. Q., and Archibald, D. D. (2013). “Quantification of cellulose nanowhiskers sulfate esterification levels,” Carbohydrate Polymers 92(2), 1809-1816. DOI: 10.1016/j.carbpol.2012.10.078
Habibi, Y., Chanzy, H., and Vignon, M. R. (2006). “TEMPO-mediated surface oxidation of cellulose whiskers,” Cellulose 13(6), 679-687. DOI: 10.1007/s10570-006-9075-y
Habibi, Y. (2014). “Key advances in the chemical modification of nanocelluloses,” Chemical Society Reviews 43(5), 1519-1542. DOI: 10.1039/C3CS60204D
Hassan, M. L., Hassan, E. A., and Oksman, K. N. (2011). “Effect of pretreatment of bagasse fibers on the properties of chitosan/microfibrillated cellulose nanocomposites,” Journal of Materials Science 46(6), 1732-1740. DOI: 10.1007/s10853-010-4992-4
Huan, S., Bai, L., Cheng, W., and Han, G. (2016). “Manufacture of electrospun all-aqueous poly (vinyl alcohol)/cellulose nanocrystal composite nanofibrous mats with enhanced properties through controlling fibers arrangement and microstructure,” Polymer 92, 25-35. DOI: 10.1016/j.polymer.2016.03.082
Huq, T., Salmieri, S., Khan, A., Khan, R. A., Le Tien, C., Riedl, B., Fraschini, C., Bouchard, J., Uribe-Calderon, J., Kamal, M. R., and Lacroix, M. (2012). “Nanocrystalline cellulose (NCC) reinforced alginate based biodegradable nanocomposite film,” Carbohydrate Polymers 90(4), 1757-1763. DOI: 10.1016/j.carbpol.2012.07.065
Isogai, A., Saito, T., and Fukuzumi, H. (2011). “TEMPO-oxidized cellulose nanofibers,” Nanoscale 3(1), 71-85. DOI: 10.1039/c0nr00583e
Jonoobi, M., Oladi, R., Davoudpour, Y., Oksman, K., Dufresne, A., Hamzeh, Y., and Davoodi, R. (2015). “Different preparation methods and properties of nanostructured cellulose from various natural resources and residues: A review,” Cellulose 22(2), 935-969. DOI: 10.1007/s10570-015-0551-0
Li, S.-M., Jia, N., Ma, M.-G., Zhang, Z., Liu, Q.-H., and Sun, R.-C. (2011). “Cellulose–silver nanocomposites: Microwave-assisted synthesis, characterization, their thermal stability, and antimicrobial property,” Carbohydrate Polymers 86(2), 441-447. DOI: 10.1016/j.carbpol.2011.04.060
Lin, N., Bruzzese, C. C., and Dufresne, A. (2012a). “TEMPO-oxidized nanocellulose participating as crosslinking aid for alginate-based sponges,” ACS Applied Materials and Interfaces 4(9), 4948-4959. DOI: 10.1021/am301325r
Lin, N., Huang, J., and Dufresne, A. (2012b). “Preparation, properties and applications of polysaccharide nanocrystals in advanced functional nanomaterials: A review,” Nanoscale 4(11), 3274-3294. DOI: 10.1039/C2NR30260H
Lu, J., and Drzal, L.T. (2010). “Microfibrillated cellulose/cellulose acetate composites: Effect of surface treatment,” Journal of Polymer Science Part B: Polymer Physics 48(2), 153-161. DOI: 10.1002/polb.21875
Lu, J., Wang, T., and Drzal, L. T. (2008). “Preparation and properties of microfibrillated cellulose polyvinyl alcohol composite materials,” Composites Part A: Applied Science and Manufacturing 39(5), 738-746. DOI: 10.1016/j.compositesa.2008.02.003
Miao, X., Lin, J., Tian, F., Li, X., Bian, F., and Wang, J. (2016). “Cellulose nanofibrils extracted from the byproduct of cotton plant,” Carbohydrate Polymers 136(20), 841-850. DOI: 10.1016/j.carbpol.2015.09.056
Montanari, S., Roumani, M., Heux, L., and Vignon, M. R. (2005). “Topochemistry of carboxylated cellulose nanocrystals resulting from TEMPO-mediated oxidation,” Macromolecules 38(5), 1665-1671. DOI: 10.1021/ma048396c
Nordqvist, D., Idermark, J., Hedenqvist, M. S., Gällstedt, M., Ankerfors, M., and Lindström, T. (2007). “Enhancement of the wet properties of transparent chitosan− acetic-acid−salt films using microfibrillated cellulose,” Biomacromolecules 8(8), 2398-2403. DOI: 10.1021/bm070246x
Parikh, A., and Madamwar, D. (2006). “Partial characterization of extracellular polysaccharides from cyanobacteria,” Bioresource Technology 97(15), 1822-1827. DOI: 10.1016/j.biortech.2005.09.008
Pawar, S. N., and Edgar, K. J. (2012). “Alginate derivatization: A review of chemistry, properties and applications,” Biomaterials 33(11), 3279-3305. DOI: 10.1016/j.biomaterials.2012.01.007
Plackett, D., Anturi, H., Hedenqvist, M., Ankerfors, M., Gällstedt, M., Lindström, T., and Siró, I. (2010). “Physical properties and morphology of films prepared from microfibrillated cellulose and microfibrillated cellulose in combination with amylopectin,” Journal of Applied Polymer Science 117(6), 3601-3609. DOI: 10.1002/app.32254
Qin, Y. (2008). “Alginate fibres: An overview of the production processes and applications in wound management,” Polymer International 57(2), 171-180. DOI: 10.1002/pi.2296
Segal, L., Creely, J. J., Martin, A. E., and Conrad, C. M. (1959). “An empirical method for estimating the degree of crystallinity of native cellulose using the X-ray diffractometer,” Textile Research Journal 29(10), 786-794. DOI: 10.1177/004051755902901003
Sellimi, S., Younes, I., Ayed, H. B., Maalej, H., Montero, V., Rinaudo, M., Dahia, M., Mechichi, T., Hajji, M., and Nasri, M. (2015). “Structural, physicochemical and antioxidant properties of sodium alginate isolated from a Tunisian brown seaweed,” International Journal Biological Macromolecules 72, 1358-1367. DOI: 10.1016/j.ijbiomac.2014.10.016
Şen, M. (2011). “Effects of molecular weight and ratio of guluronic acid to mannuronic acid on the antioxidant properties of sodium alginate fractions prepared by radiation-induced degradation,” Applied Radiation and Isotopes 69(1), 126-129. DOI: 10.1016/j.apradiso.2010.08.017
Sibaja, B., Culbertson, E., Marshall, P., Boy, R., Broughton, R. M., Solano, A. A., Esquivel, M., Parker, J., De La Fuente, L., and Auad, M. L. (2015). “Preparation of alginate–chitosan fibers with potential biomedical applications,” Carbohydrate Polymers 134 598-608. DOI: 10.1016/j.carbpol.2015.07.076
Siró, I., and Plackett, D. (2010). “Microfibrillated cellulose and new nanocomposite materials: A review,” Cellulose 17(3), 459-494. DOI: 10.1007/s10570-010-9405-y
Soares, J. P., Santos, J. E., Chierice, G. O., and Cavalheiro, E. T. G. (2004). “Thermal behavior of alginic acid and its sodium salt,” Eclética Química 29(2), 57-64. DOI: 10.1590/S0100-46702004000200009
Stojanovic, R., Belscak-Cvitanovic, A., Manojlovic, V., Komes, D., Nedovic, V., and Bugarski, B. (2012). “Encapsulation of thyme (Thymus serpyllum L.) aqueous extract in calcium alginate beads,” Journal of the Science of Food and Agriculture 92(3), 685-696. DOI: 10.1002/jsfa.4632
Ureña-Benavides, E. E., Brown, P. J., and Kitchens, C. L. (2010). “Effect of jet stretch and particle load on cellulose nanocrystal−alginate nanocomposite fibers,” Langmuir 26(17), 14263-14270. DOI: 10.1021/la102216v
Watthanaphanit, A., Supaphol, P., Tamura, H., Tokura, S., and Rujiravanit, R. (2010). “Wet-spun alginate/chitosan whiskers nanocomposite fibers: Preparation, characterization and release characteristic of the whiskers,” Carbohydrate Polymers 79(3), 738-746. DOI: 10.1016/j.carbpol.2009.09.031
Yang, G., Zhang, L., Peng, T., and Zhong, W. (2000). “Effects of Ca2+ bridge cross-linking on structure and pervaporation of cellulose/alginate blend membranes,” Journal of Membrane Science 175(1), 53-60. DOI: 10.1016/S0376-7388(00)00407-5
Zhao, X., Li, Q., Ma, X., Quan, F., Wang, J., and Xia, Y. (2015). “The preparation of alginate–AgNPs composite fiber with green approach and its antibacterial activity,” Journal of Industrial Engineering Chemistry 24, 188-195. DOI: 10.1016/j.jiec.2014.09.028
Zhou, C., Chu, R., Wu, R., and Wu, Q. (2011). “Electrospun polyethylene oxide/cellulose nanocrystal composite nanofibrous mats with homogeneous and heterogeneous microstructures,” Biomacromolecules 12(7), 2617-2625. DOI: 10.1021/bm200401p
Article submitted: May 19, 2017; Peer review completed: September 9, 2017; Revised version received and accepted: September 12, 2017; Published: September 19, 2017.
DOI: 10.15376/biores.12.4.8180-8198