
Abstract
The influence of elevated temperatures on mechanical behavior was studied for curaua, hemp, and sisal natural fibers. Tensile tests were performed on fibers heated at 100 °C, 150 °C, and 200 °C for 24 h, and reference samples were maintained without thermal treatment for comparisons. The cross sectional area of the fibers was measured using a scanning electron microscope (SEM), and the image analysis was performed using the open source software Fiji/ImageJ. These data allowed the computation of the tensile stresses and the correlation of the fiber morphology with its macro-mechanical behavior. The thermal degradation behavior of the natural fibers was measured via thermo-gravimetric analysis (TGA) and X-ray diffraction (XRD). The morphological and mechanical characteristics were described and discussed on a microstructural basis. The results showed that the loss of moisture leads to a significant increase in tensile strength before reaching the limits of the degradation range.
Download PDF
Full Article
Degradation Mechanisms of Curaua, Hemp, and Sisal Fibers Exposed to Elevated Temperatures
Felipe Pinheiro Teixeira,a Otávio da Fonseca Martins Gomes,b,c and Flávio de Andrade Silva a,*
The influence of elevated temperatures on mechanical behavior was studied for curaua, hemp, and sisal natural fibers. Tensile tests were performed on fibers heated at 100 °C, 150 °C, and 200 °C for 24 h, and reference samples were maintained without thermal treatment for comparisons. The cross sectional area of the fibers was measured using a scanning electron microscope (SEM), and the image analysis was performed using the open source software Fiji/ImageJ. These data allowed the computation of the tensile stresses and the correlation of the fiber morphology with its macro-mechanical behavior. The thermal degradation behavior of the natural fibers was measured via thermo-gravimetric analysis (TGA) and X-ray diffraction (XRD). The morphological and mechanical characteristics were described and discussed on a microstructural basis. The results showed that the loss of moisture leads to a significant increase in tensile strength before reaching the limits of the degradation range.
Keywords: Natural fibers; Elevated temperatures; Morphology; Mechanical behavior
Contact information: a: Civil and Environmental Engineering Department, Pontifícia Universidade Católica do Rio de Janeiro (PUC-Rio), 22451-900, Rio de Janeiro – RJ, Brazil; b: Centre for Mineral Technology (CETEM), Av. Pedro Calmon, 900, Cidade Universitária, Rio de Janeiro 21941-908, Brazil; c: Postgraduate Program in Geosciences, National Museum, Federal University of Rio de Janeiro (UFRJ), Av. Quinta da Boa Vista, S/N, Bairro Imperial de São Cristóvão, Rio de Janeiro, 20940-040, Brazil;
* Corresponding author: fsilva@puc-rio.br
INTRODUCTION
Research on the addition of natural fibers to engineering materials has been widespread, seeking environmentally friendly and energy-efficient solutions. Natural fibers are particularly interesting reinforcing components for composites, as they are biodegradable, harmless to the environment, and economically more viable than synthetic fibers. Therefore, the mechanical, morphological, and durability characteristics of fibers such as abaca leaf, cotton, curaua, flax, hemp, jute, sisal, etc., have been investigated (Kicińska-Jakubowska et al. 2012; Komuraiah et al. 2014; Yan et al. 2016).
Because these fibers are natural materials, they present morphological variety, which directly influences the tensile strength and Young’s modulus. Disparities among measured values may be associated with the production efficiency, natural conditions, extraction methods, and processing conditions of the fiber. These variations in the mechanical behavior can be described by morphological characteristics.
The morphologies of curaua and sisal fibers have been evaluated, correlating their mechanical properties via image analysis (Fidelis et al. 2013). These fibers presented tensile strengths greater than 249 MPa and Young’s moduli greater than 19 GPa, classifying them as high-performance fibers. The highest tensile strength was observed for curaua, at 543 MPa with a Young’s modulus around 64 GPa, followed by sisal, at 484 MPa and 19 GPa, respectively. The correlation of morphology (by the real cross-section) with the mechanical properties showed that as the internal area of the lumens decreases and the secondary cell wall thickness increases, the fiber strength and Young’s modulus increase.
Analysis of the influence of stem sampling area on the mechanical properties of hemp fibers showed that fibers from the middle of the stem exhibit the greatest ultimate strength and elongation, with the Young’s modulus around 19 GPa and the tensile strength not exceeding 500 MPa (Duval et al. 2011). Compared to bottom and top sampling areas, the ultimate tensile strength differences were approximately 23% and 18%, respectively. Liu et al. (2015) showed that hemp fibers from the middle section also exhibited the best mechanical performance, presenting the highest ultimate tensile strength and strain of 842 MPa and 5.7%, respectively, and a moderate stiffness of 28.8 GPa. Fibers from the top section showed moderate mechanical properties, with an ultimate tensile strength of 809 MPa, a strain of 4.7%, and the highest stiffness, of 31.5 GPa. The variation in mechanical properties of fibers with stem section is explained by the differences in morphological features and composition.
In natural fibers, thermal degradation involves two main steps. The first is the thermal depolymerization of the hemicellulose and the cleavage of glycosidic linkages of cellulose. The second is related to the decomposition of the α-cellulose. The decomposition of lignin occurs at temperatures between 200 °C and 500 °C (Manfredi et al. 2006). In derivative thermogravimetric (DTG) analysis of sisal fibers, the decomposition starts at 215 °C, the decomposition of the hemicellulose happens at 290 °C, and the maximum degradation rate occurs during α-cellulose decomposition at 340 °C. Yao et al. (2008) analyzed the thermal decomposition process of 10 types of natural fibers, included wood, bamboo, agricultural residue and bast fibers. The thermal decomposition process resulted in a similar TG and DTG curves due to being lignin cellulosic material. It was noticed an onset decomposition temperature about 215 °C for most of the natural fibers and weight loss in this range was observed around 5%. The maximum decomposition rate of main natural fibers happened about 290 °C, except for maple and pine (an average of 310 °C). These same ranges of decomposition were also presented by other authors (Yang et al. 2007; Collard and Blin 2014; Ovalle-Serrano et al. 2017; Yu et al. 2017).
The thermal characterization of sisal fibers by thermogravimetric analysis (TGA) presented a mass loss of approximately 3% from 30 °C to 220 °C, attributed to water loss in fibers (intramolecular and intermolecular dehydration reactions) (Martin et al. 2009). The fibers’ degradation begins around 250 °C and continues quickly as temperature increases, having a mass loss of 72% at approximately 420 °C. Due to the slow decomposition of residues, the mass loss from 420 °C to approximately 520 °C is only 5%. After 520 °C, the ash content is around 20%.
The thermo-mechanical behavior of hemp fibers at temperatures between 20 °C and 200 °C showed an activation of their visco-elastic properties, corresponding to relaxation of the constituent polymers (hemicellulose and lignin), and decreases in rigidity and endurance, attributed to thermal degradation of the cellular walls, at temperatures between 150 °C and 180 °C (Placet 2009).The literature presents extensive results about the thermal degradation of cellulose-based structures, especially through thermo-gravimetrical analysis, showing indices of moisture loss, decomposition and maximum weight loss until reaching the limits of the degradation range. However, such analyzes do not represent how these levels of thermal degradation can truly influence the mechanical behavior of exposed natural fibers. The present work investigated the mechanics of three different natural fibers (sisal, curaua, and hemp) and how elevated temperatures can influence their mechanical behavior. The morphologies and cross-sectional areas of each fiber were measured using a scanning electron microscope (SEM) and an image analysis routine. The different fibers were tested under direct tension before and after being exposed at 100 °C, 150 °C, and 200 °C. The mechanisms of degradation were explained through TGA and X-ray diffraction (XRD) analyses and by microstructural observation.
EXPERIMENTAL
Natural Fibers
The curaua fibers were provided by the Pematec Company (Santarém, Pará, Brazil). The fibers were obtained in the Amazon region, extracted from the Ananas erectifolius plant by the mechanical process of decortication (Silva et al. 2008; Fidelis et al. 2013). The hemp fibers were obtained from Unipak A/S (Galten, Denmark), which manufactures skeins made of natural hemp for the plumbing industry. The sisal fibers were obtained from farms located in Valente, Bahia, Brazil. They were extracted from the leaf of the Agave sisalana plant by the same process used for the curaua fiber.
All the fibers were first treated with hot water (approximately 70 °C) for 1 h. This procedure aimed to eliminate the maximum amount of impurities retained on the fiber surface. Thereafter, the fibers were air dried for 48 h.
Mechanical Tests
For the tensile tests, the dried specimens were arranged according to ASTM C1557-14 (2014) with gauge lengths corresponding to 20 mm. The fibers were fixed in kraft paper tabs (140 g/cm²) and then heated at 100 °C, 150 °C, and 200 °C for 24 h. Reference samples were maintained without thermal treatment (room at approximately 22 °C and 60% relative humidity) for comparison. The specimens were subjected to tensile loading performed in a servo hydraulic MTS 810 system with a 100 N load cell and an external LVDT (MTS, Eden Prairie, MN, USA) to achieve greater accuracy in data acquisition. Fifteen specimens of fiber were tested for each group (reference, 100 °C, 150 °C, and 200 °C) and the tensile tests were carried on under displacement control at a rate of 0.1 mm/min. All tests were performed at room temperature (approximately 22 °C) with a relative humidity of approximately 60%.
Microstructural Investigation
The fiber microstructure was investigated using an FEI Quanta 400 scanning electron microscope (Thermo Fisher Scientific, Hillsboro, OR, USA). For the dimensional analysis, the samples were prepared by cold embedding in epoxy resin (Gomes et al. 2018). A solid resin block 30 mm in diameter and approximately 12 mm in height was drilled with a 1.5 mm diameter drill to create 15 holes, in which 10 mm length fiber samples were carefully placed, as illustrated in Fig. 1(a). Thereafter, the holes with fibers were filled with more epoxy resin, and a vacuum pump was used to ensure no bubbles were formed during its curing. After curing, the block was ground and polished in an automatic polishing machine (Struers Tegramin 20, Cleveland, OH, USA). The grinding was performed with 125 µm, 40 µm, 9 µm, and 6 µm sized diamond-particle-impregnated metal discs, for 3 min, 4 min, 4 min, and 10 min, respectively. Then, the blocks were polished using 3 μm and 1 µm diamond suspensions for approximately 20 min each to produce a high flatness surface. Subsequently, the blocks were covered with evaporated carbon to become conductive, as shown in Fig. 1(b). This preparation procedure can provide a suitable contrast in SEM images to analyze the fiber microstructure. The image analysis was carried out using the open source software Fiji/ImageJ (Schindelin et al. 2012; Schneider et al. 2012). A contour line was interactively drawn to delineate fiber cross-sections, and their areas were then measured.
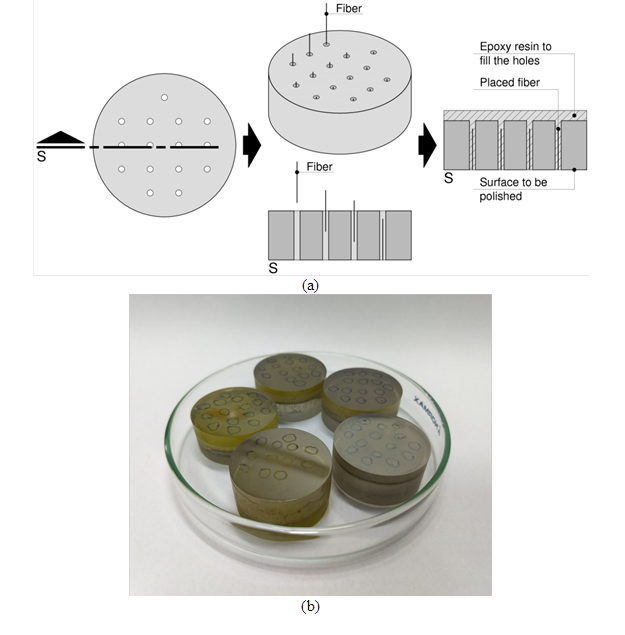
Fig. 1. The sample preparation procedure: (a) schematic illustration of the block where fiber samples were placed and embedded within epoxy resin and (b) the blocks covered with evaporated carbon, to become conductive and provide suitable contrast in SEM
TG and XRD Analyses
The thermogravimetric analysis (TGA) was performed under N2 atmosphere, from 25 °C up to 500 °C, using a TA Instruments (New Castle, DE, USA) SDT Q600. Approximately 10 mg of each sample was used, and the analysis was conducted at a heating rate of 5 °C/min.
The XRD measurements were performed on a Bruker D8 set to 40 kV and 25 mA using CuKα radiation (λ = 1.5406 nm). The profiles were recorded in an angular range 2θ from 5° up to 100° with increments of 0.01°. For sample preparation, the fibers were scissor cut to particle sizes of less than 0.5 mm. Four samples by fiber type were analyzed for each group (reference, 100 °C, 150 °C and 200 °C). To determine the amount of crystallinity, the deconvolution method of crystalline peaks and amorphous halo was used. For this process, OriginPro8 software (OriginLab Corp., Northampton, USA) with a Gaussian function was used. The diffractogram was separated into various components that independently contributed to the formation of the peaks in each phase. The quantitative decomposition of XRD provided a measure of crystallinity index using Eq. 1,
 (1)
(1)
where CI is the crystallinity index of the material, Ac is the area of crystalline peaks, and Aa is the area of amorphous halo.
In order to evaluate the statistical significance of CI variations, the two-sample t-test was used as the statistical method. The parameter α = 0.05 was set as the risk level in all statistical analysis, and p < 0.05 was considered to be statistically significant.
RESULTS AND DISCUSSION
Fiber Morphology
Hierarchically, in a simplified way, a single natural fiber is formed by several microfibers that have their cell walls (primary cell wall and secondary wall divided into three layers around the lumen) constituted by a series of helically wound cellular microfibrils formed from cellulose molecules (Zimmermann et al. 2004; John and Thomas 2008; Wegst et al. 2015). The studied fibers showed different morphologies when analyzed by dimensional parameters. The geometries of the cross sections were also distinct, and these variations were related to the quantity, size, and organization of the microfibers. Table 1 summarizes the morphological characteristics of the curaua, hemp, and sisal fibers.
Table 1. Morphological Characteristics of Curaua, Hemp, and Sisal Fibers

The cross section of the curaua fiber (Fig. 2(a)) presented a star shape with area around 0.006 mm² and a large amount of microfibers in its structure, approximately 404 per section, the greatest amount of microfibers among the three species studied. These microfibers presented an average area of 18 μm² and cell wall thickness of 1.6 μm, the smallest area and cell wall thickness of microfibers among the three species studied. The hemp fiber (Fig. 2(b)) showed the smallest area among the three fibers, measuring 0.002 mm², and could not conclusively be associated with a specific form due to their average amount of microfibers per section being around 11, which together showed a relatively uniform geometry of agglomeration. However, their microfibers presented an area around 224 μm² with the cell wall thickness around 6.2 μm, the largest among the three fibers studied. The sisal fiber showed the largest cross section area, compared with curaua and hemp, measuring 0.030 mm². The sisal fiber (Fig. 2(c)) presented an arched shape (Silva et al. 2011), with approximately 228 microfibers, measuring about 137μm² in area with a cell wall thickness around 2.9 μm.

Fig. 2. Cross sections of curaua (a), hemp (b), and sisal (c) fibers by SEM
Fidelis et al. (2013) also investigated the morphological characteristics of curaua and sisal fibers, as showed in Table 2. Some of these results, such as the amount of microfibers per cross section, presented a wide variation: a reduction of 97% and 39% for curaua and sisal, respectively. However, even with this large rate difference, the cross-sectional area results can be considered close. These variations can be explained by the morphological disuniformity presented by the natural fibers structure, which provides different characteristics, including the mechanical ones, due the influence of the morphology variation of fibers from stem’s top, middle and bottom (Duval et al. 2011; Liu et al. 2015).
Table 2. Natural Fibers Morphological Characteristics From Literature

When exposed to thermal treatments, the fibers showed different levels of degradation. The curaua and hemp fibers (Figs. 3 and 4) exposed at 200 °C presented delamination of the microfibers, with the weakening of the middle lamellas visible, especially in the hemp fibers that suffered total bond collapse.

Fig. 3. The curaua reference (a) and treated at 200 °C (b)

Fig. 4. The hemp reference (a) and treated at 200 °C (b)
The sisal fibers (Fig. 5) at 200 °C showed the greatest thermal resistance and did not show any signs of weakening. The high thermal resistance of sisal in comparison to curaua and hemp can be explained by the amount of hemicelluloses—curaua and hemp have hemicellulose amounts around 20% to 23% (Komuraiah et al. 2014; Ferreira et al. 2015b; Yan et al. 2016) but sisal only 10% (Sydenstricker et al. 2003; Silva et al. 2008)—which is responsible for most of the thermal degradation behavior of natural fibers (Beg and Pickering 2008; Ornaghi et al. 2014).

Fig. 5. The sisal reference (a) and treated at 200 °C (b)
TG and XRD Analysis
The TGA of curaua, hemp, and sisal fibers showed similar patterns of weight loss (Fig. 6). Up to 100 °C, the samples showed a moisture decrease, represented by a weight loss of approximately 9%, and then stabilized from that point to 200 °C. From there to approximately 290 °C the hemicellulose degradation occurred and thereafter the thermal decomposition of cellulose remains up to approximately 350 °C, as also presented by many other authors (Alvarez and Vázquez 2004; Manfredi et al. 2006; Martin et al. 2009; Krishnaiah et al. 2017). At this point, the thermal degradation caused weight losses of 78%, 67%, and 72% for curaua, hemp, and sisal, respectively. Over 450 °C the continuous weight loss is due to oxidation of remnant carbon (Ovalle-Serrano et al. 2018).
According with DTG analyses, the maximum weight loss was observed at 329 °C for hemp, 336 °C for curaua, and 342 °C for sisal, which is consistent with that observed in the XRD analysis where sisal fibers also showed higher thermal stability, probably due the lower content of hemicelluloses when compared to curaua and hemp fibers. The range from 100 °C to 200 °C by TGA and DTG showed no significant changes occurring with these fibers (beyond those already shown up to 100 °C).
The XRD analysis of curaua, hemp, and sisal fibers was performed for the reference and 200 °C treated samples, and the deconvolution of crystalline peaks and amorphous halo was performed in the range of 5° to 60° (2θ). The highest intensity peak in all cases was noticed at 2θ between 22.0° and 22.6° reflection assigned to the (002) crystallographic plane. The average intensity peak was observed at 2θ between 14.5° and15.3 ° (101), with the lowest intensity peak at 2θ between 33.9° and 34.1° (040).
Only the hemp samples presented prominent peaks at 2θ between 15.9° and 16.2°  . These results are compatible with other analyses about the crystalline structure of cellulose (Park et al. 2010; Poletto et al. 2014; Ferreira et al. 2015a; Diaz et al. 2016).
. These results are compatible with other analyses about the crystalline structure of cellulose (Park et al. 2010; Poletto et al. 2014; Ferreira et al. 2015a; Diaz et al. 2016).

Fig. 6. TGA and DTG of curaua, hemp, and sisal fibers
The peaks at 2θ from 33.9° to 34.1° (040) were not included in the deconvolution analysis due to their very low intensity (Diaz et al. 2016) (Fig. 7). Table 3 presents the natural fibers’ crystallinity indexes.
Table 3. Crystallinity Indexes of Curaua, Hemp, and Sisal Fibers, Reference and after Exposure at 200 °C
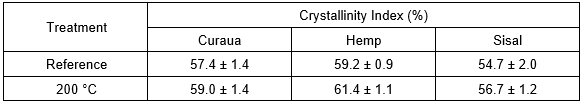
Comparing the reference fibers with those exposed at 200 °C, the CI presented increases of about 3%, 4%, and 4% for curaua, hemp, and sisal, respectively. The crystallinity degree of cellulose is directly related to the rigidity and flexibility of the natural fibers: their rigidity increases, and flexibility decreases, with increasing ratio of crystalline to amorphous regions (Poletto et al. 2014). In this case, however, the CI increases at 200 °C probably were associated with the loss of humidity (at 100 °C). For curaua and sisal fibers, this difference in crystallinity index values between the reference and 200 °C samples was not statistically significant, as described by the p-values in Table 4. However, the hemp fibers presented p < 0.05, which indicates there was statistical significance concerning its variation.

Fig. 7. The XRD and software analysis to determinate the CI of curaua reference (a), curaua exposed to 200 °C (b), hemp reference (c), hemp exposed to 200 °C (d), sisal reference (e), and sisal exposed to 200 °C (f) (a.u. stands for arbitrary units)
Table 4. Statistical Significance of Crystallinity Index Values Between the Reference and 200 °C Samples by Two-Sample T-Test

In this regard, other authors (Puangsin et al. 2013; Ovalle-Serrano et al. 2018) reported a decrease about 10 % in CI of bleached natural fibers and affirmed that these variations does not affect significantly the biopolymer properties, however, these kinds of chemical results must be confronted with experimental mechanical analyzes.
Fiber Mechanical Behavior
The mechanical behaviors of all the fibers under all conditions are summarized in Table 5. The highest tensile strength among reference samples was observed for curaua (760 MPa), followed by the hemp (480 MPa) and sisal (357 MPa).
Table 5. Tensile Tests Results for Curaua, Hemp, and Sisal Fibers

The curaua and hemp fibers presented similar mechanical behaviors, both showing a high stiffness, while sisal tended to be more ductile, as shown in Fig. 8(a). This is probably related to their microfibrillar angle (hemp approximately 6° and sisal between 10° and 22°) (Dittenber and GangaRao 2012; Komuraiah et al. 2014; Yan et al. 2016), where smaller angles lead to greater strength and stiffness and larger angles yield greater ductility (Azwa et al. 2013). In sisal fibers, a non-linear region at the initial portion of the curve can be explained as a collapse of the weak primary cell walls and delamination between microfibers (Silva et al. 2008).
Figure 8(b) presents the relation between tensile strength and temperature from references up to 150 °C treated samples. In comparison with the reference samples, all studied fibers presented tensile strength gains when subjected to 100 °C for 24 h (Fig. 8(b)). The curaua, hemp, and sisal fibers at 100 °C showed tensile strength gains of approximately 94%, 18%, and 6%, respectively. This result shows that temperatures around 100 °C do not compromise the tensile behavior of natural fibers but can admit mechanical gains by the loss of humidity.

Fig. 8. Mechanical behavior of reference curaua, hemp, and sisal fibers (a) and the relation between tensile strength and temperature from references up to 150 °C treated samples (b)
Figure 9 compares the fibers’ tensile behaviors when thermally treated. At 100 °C, the curaua and sisal fibers presented Young’s modulus increases of approximately 34% and 30%, respectively. Those fibers also presented increases in stiffness, while the hemp fibers exhibited a decrease of about 24% after being exposed at 100 °C. These variations in stiffness can be attributed to rearrangements and reorientations of the cellulose microfibrils and/or changes in the crystallinity fraction that may occur in the fibers (Placet 2009).
At 150 °C, all fibers started showing losses in tensile strength. The curaua and sisal fibers showed values below their respective reference samples. The loss showed by the hemp fibers at 150 °C was not sufficient to place it below the reference samples, but at 200 °C these were the most damaged, presenting no mechanical resistance and easily breaking if handled, which agrees with the results of the statistical significance.
At 200 °C, curaua and sisal fibers had drastic decreases in tensile strength, around 51 MPa and 38 MPa, respectively (Fig. 9(d)). The thermal treatment at 200 °C completely damaged the fibers, which is attributable to the thermal degradation of the cellular walls at temperatures between 150 °C and 180 °C (Placet 2009). The same occurred with thermal treatments on kenaf bast fibers at 170 °C and 180 °C for 24 h in other experiments (Xue et al. 2009).
For the sisal fibers (Fig. 9(c)), the non-linear region occurring after 50 MPa gradually disappeared as the temperature increased. The changes at the initial non-linear region may be associated with the loss of humidity in those thermal ranges, which can increase the stiffness of the fiber and, in this case, probably opposes the effect of the previously described delamination of the microfibers (Silva et al. 2008).
Comparing the mechanical results with the TG and XRD analyses, weight loss and CI can be described as stable in the range from 100 °C to 200 °C, but the mechanical degradation is notable. In the range between 100 °C and 150 °C, partial decreases in tensile behavior occur. From there to 200 °C, the fibers are already mechanically compromised.

Fig. 9. Comparison of curaua (a), hemp (b), and sisal (c) fibers’ tensile behaviors when thermally treated; the curaua and sisal fibers at 200 °C (d)
CONCLUSIONS
- Although similar at first, the curaua, hemp, and sisal fibers showed distinct morphologies. Characteristics such as cross section area and amount of microfibers per section evinced dimensional variability in microstructure, which influences mechanical behavior.
- When exposed at 100 °C for 24 h, all fibers presented tensile strength improvements, especially the curaua fibers, which showed an increase of approximately 94%, followed by hemp and sisal, with increases of approximately 18% and 6%, respectively.
- The curaua and sisal fibers also showed Young’s modulus increases at 100 °C, presenting stiffness increases, while the hemp fibers exhibited a decrease in Young’s modulus.
- The behavior of all studied fibers presented on TG and XRD analysis for the range from 100 °C to 200 °C is described as stable, but their mechanical behavior showed severe degradation within the range from 150 °C to 200 °C. In this case the mechanical degradation can be traced back to the loss of bond among the several microfibers that compose the fibers.
- All fibers exposed at 200 °C for 24 h became fragile and brittle, presenting drastic decreases in mechanical resistance. The curaua and hemp fibers showed clear microfiber delamination, especially the hemp, which suffered total bond collapse. The sisal fibers showed the greatest thermal resistance and did not show signs of weakening. Nevertheless, the sisal fiber mechanical bearing capacity was also compromised after 200 °C.
- The overall analysis of elevated temperatures on the mechanical behavior of natural fibers showed that, in three cases with three distinct species (curaua, hemp, and sisal), heating up to 100 °C did not compromise their tensile behavior but could admit mechanical gains by the loss of moisture content.
ACKNOWLEDGMENTS
The authors acknowledge support from the Brazilian funding agencies CNPq, CAPES, FINEP, and FAPERJ. They also thank LCPMA-UERJ and Professor Fátima Zotin for collaboration with TGA. F.P. Teixeira thanks CAPES for the D.Sc. candidate scholarship number 88887.165362/2018-00.
REFERENCES CITED
Alvarez, V. A., and Vázquez, A. (2004). “Thermal degradation of cellulose derivatives/starch blends and sisal fibre biocomposites,” Polymer Degradation and Stability 84(1), 13-21. DOI: 10.1016/j.polymdegradstab.2003.09.003
ASTM C1557-14 (2014). “Standard test method for tensile strength and Young’s modulus of fibers,” ASTM International, West Conshohocken, USA.
Azwa, Z. N., Yousif, B. F., Manalo, A. C., and Karunasena, W. (2013). “A review on the degradability of polymeric composites based on natural fibres,” Materials and Design 47, 424-442. DOI: 10.1016/j.matdes.2012.11.025
Beg, M. D. H., and Pickering, K. L. (2008). “Accelerated weathering of unbleached and bleached kraft wood fibre reinforced polypropylene composites,” Polymer Degradation and Stability 93(10), 1939-1946. DOI: 10.1016/j.polymdegradstab.2008.06.012
Collard, F., and Blin, J. (2014). “A review on pyrolysis of biomass constituents : Mechanisms and composition of the products obtained from the conversion of cellulose, hemicelluloses and lignin,” Renewable and Sustainable Energy Reviews 38, 594-608. DOI: 10.1016/j.rser.2014.06.013
Diaz, J. P. V., Silva, F. A., and d’Almeida, J. R. M. (2016). “Effect of peach palm fiber microstructure on its tensile behavior,” BioResources 11(4), 10140-10157. DOI: 10.15376/biores.11.4.10140-10157
Dittenber, D. B., and GangaRao, H. V. S. (2012). “Critical review of recent publications on use of natural composites in infrastructure,” Composites Part A: Applied Science and Manufacturing 43(8), 1419-1429. DOI: 10.1016/j.compositesa.2011.11.019
Duval, A., Bourmaud, A., Augier, L., and Baley, C. (2011). “Influence of the sampling area of the stem on the mechanical properties of hemp fibers,” Materials Letters 65(4), 797-800. DOI: 10.1016/j.matlet.2010.11.053
Ferreira, S. R., Silva, F. D. A., Lima, P. R. L., and Toledo Filho, R. D. (2015a). “Effect of fiber treatments on the sisal fiber properties and fiber–matrix bond in cement based systems,” Construction and Building Materials 101, 730-740. DOI: 10.1016/j.conbuildmat.2015.10.120
Ferreira, S. R., Silva, F. de A., Lima, P. R. L., and Toledo Filho, R. D. (2015b). “Effect of natural fiber hornification on the fiber matrix interface in cement based composite systems,” Key Engineering Materials 668, 118-125. DOI: 10.4028/www.scientific.net/KEM.668.118
Fidelis, M. E., Pereira, T. V. C., Gomes, O. D. F. M., De Andrade Silva, F., and Toledo Filho, R. D. (2013). “The effect of fiber morphology on the tensile strength of natural fibers,” Journal of Materials Research and Technology 2(2), 149-157. DOI: 10.1016/j.jmrt.2013.02.003
Gomes, O. da F. M., Teixeira, F. P., Lima, J. F., and Silva, F. de A. (2018). “On the measurement of cross-sectional area of natural fibers,” in: 4th Brazilian Conference on Composite Materials, Rio de Janeiro, Brazil, 1-8. DOI: 10.21452/bccm4.2018.10.10
John, M. J., and Thomas, S. (2008). “Biofibres and biocomposites,” Carbohydrate Polymers 71(3), 343-364. DOI: 10.1016/j.carbpol.2007.05.040
Kicińska-Jakubowska, A., Bogacz, E., and Zimniewska, M. (2012). “Review of natural fibers. Part I—Vegetable fibers,” Journal of Natural Fibers 9(3), 150-167. DOI: 10.1080/15440478.2012.703370
Komuraiah, A., Kumar, N. S., and Prasad, B. D. (2014). “Chemical composition of natural fibers and its influence on their mechanical properties,” Mechanics of Composite Materials50(3), 359-376. DOI: 10.1007/s11029-014-9422-2
Krishnaiah, P., Ratnam, C. T., and Manickam, S. (2017). “Enhancements in crystallinity, thermal stability, tensile modulus and strength of sisal fibres and their PP composites induced by the synergistic effects of alkali and high intensity ultrasound (HIU) treatments,” Ultrasonics Sonochemistry 34, 729-742. DOI: 10.1016/j.ultsonch.2016.07.008
Liu, M., Fernando, D., Meyer, A. S., Madsen, B., Daniel, G., and Thygesen, A. (2015). “Characterization and biological depectinization of hemp fibers originating from different stem sections,” Industrial Crops and Products 76, 880-891. DOI: 10.1016/j.indcrop.2015.07.046
Manfredi, L. B., Rodríguez, E. S., Wladyka-Przybylak, M., and Vázquez, A. (2006). “Thermal degradation and fire resistance of unsaturated polyester, modified acrylic resins and their composites with natural fibres,” Polymer Degradation and Stability 91(2), 255-261. DOI: 10.1016/j.polymdegradstab.2005.05.003
Martin, A. R., Martins, M. A., Mattoso, L. H. C., and Silva, O. R. R. F. (2009). “Caracterização química e estrutural de fibra de sisal da variedade Agave sisalana,” Polímeros 19(1), 40-46. DOI: 10.1590/S0104-14282009000100011
Ornaghi, H. L., Poletto, M., Zattera, A. J., and Amico, S. C. (2014). “Correlation of the thermal stability and the decomposition kinetics of six different vegetal fibers,” Cellulose 21(1), 177-188. DOI: 10.1007/s10570-013-0094-1
Ovalle-Serrano, S.A., Blanco-Tiradob, C., and Combariza, M.Y. (2017). “Exploring the composition of raw and delignified Colombian fique fibers, tow and pulp,” Cellulose 25, 151-165. DOI: 10.1007/s10570-017-1599-9
Ovalle-Serrano, S. A., Gómeza, F. N., Blanco-Tiradob, C., and Combariza, M. Y. (2018). “Isolation and characterization of cellulose nanofibrils from Colombian Fique decortication by-products,” Carbohydrate Polymers 189, 169-177. DOI: 10.1016/j.carbpol.2018.02.031
Park, S., Baker, J. O., Himmel, M. E., Parilla, P. A., and Johnson, D. K. (2010). “Cellulose crystallinity index: Measurement techniques and their impact on interpreting cellulase performance,” Biotechnology for Biofuels 3(10), 1-10. DOI: 10.1186/1754-6834-3-10
Placet, V. (2009). “Characterization of the thermo-mechanical behaviour of hemp fibres intended for the manufacturing of high performance composites,” Composites Part A: Applied Science and Manufacturing 40(8), 1111-1118. DOI: 10.1016/j.compositesa.2009.04.031
Poletto, M., Ornaghi Júnior, H. L., and Zattera, A. J. (2014). “Native cellulose: Structure, characterization and thermal properties,” Materials 7(9), 6105-6119. DOI: 10.3390/ma7096105
Puangsin, B., Yang, Q., Saito, T., and Isogai, A. (2013). “Comparative characterization of TEMPO-oxidized cellulose nanofibril films prepared from non-wood resources,” International Journal of Biological Macromolecules 59, 208-213. DOI: 10.1016/j.ijbiomac.2013.04.016
Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., et al. (2012). “Fiji: An open-source platform for biological-image analysis,” Nature Methods 9, 676-682. DOI: 10.1038/nmeth.2019
Schneider, C. A., Rasband, W. S., and Eliceiri, K. W. (2012). “NIH Image to ImageJ: 25 years of image analysis,” Nature Methods 9, 671-675. DOI: 10.1038/nmeth.2089
Silva, F. D. A., Mobasher, B., Soranakom, C., and Filho, R. D. T. (2011). “Effect of fiber shape and morphology on interfacial bond and cracking behaviors of sisal fiber cement based composites,” Cement and Concrete Composites 33(8), 814-823. DOI: 10.1016/j.cemconcomp.2011.05.003
Silva, F. de A., Chawla, N., and Filho, R. D. de T. (2008). “Tensile behavior of high performance natural (sisal) fibers,” Composites Science and Technology 68(15-16), 3438-3443. DOI: 10.1016/j.compscitech.2008.10.001
Silva, F. de A., Mobasher, B., and Filho, R. D. T. (2009). “Cracking mechanisms in durable sisal fiber reinforced cement composites,” Cement and Concrete Composites 31(10), 721-730. DOI: 10.1016/j.cemconcomp.2009.07.004
Sydenstricker, T. H. D., Mochnaz, S., and Amico, S. C. (2003). “Pull-out and other evaluations in sisal-reinforced polyester biocomposites,” Polymer Testing 22(4), 375-380. DOI: 10.1016/S0142-9418(02)00116-2
Wegst, U. G. K., Bai, H., Saiz, E., Tomsia, A. P., and Ritchie, R. O. (2015). “Bioinspired structural materials,” Nature Materials 14(1), 23-36. DOI: 10.1038/nmat4089
Xue, Y., Du, Y., Elder, S., Wang, K., and Zhang, J. (2009). “Temperature and loading rate effects on tensile properties of kenaf bast fiber bundles and composites,” Composites Part B: Engineering 40(3), 189-196. DOI: 10.1016/j.compositesb.2008.11.009
Yao, F., Wu, Q., Lei, Y., Guo, W., and Xu, Y. (2008). “Thermal decomposition kinetics of natural fibers: Activation energy with dynamic thermogravimetric analysis,” Polymer Degradation and Stability 93, 90-98. DOI: 10.1016/j.polymdegradstab.2007.10.012
Yan, L., Kasal, B., and Huang, L. (2016). “A review of recent research on the use of cellulosic fibres, their fibre fabric reinforced cementitious, geo-polymer and polymer composites in civil engineering,” Composites Part B: Engineering 92, 94-132. DOI: 10.1016/j.compositesb.2016.02.002
Yang, H., Yan, R., Chen, H., Lee, D. H., and Zheng, C. (2007). “Characteristics of hemicellulose, cellulose and lignin pyrolysis,” Fuel 86, 1781-1788. DOI: 10.1016/j.fuel.2006.12.013
Yu, J., Paterson, N., Blamey, J., and Millan, M. (2017). “Cellulose , xylan and lignin interactions during pyrolysis of lignocellulosic biomass,” Fuel 191, 140-149. DOI: 10.1016/j.fuel.2016.11.057
Zimmermann, T., Pöhler, E., and Geiger, T. (2004). “Cellulose fibrils for polymer reinforcement,” Advanced Engineering Materials 6(9), 754-761. DOI: 10.1002/adem.200400097
Article submitted: August 15, 2018; Peer review completed: November 18, 2018; Revised version received: December 13, 2018; Accepted: December 31, 2018; Published: January 8, 2019.
DOI: 10.15376/biores.14.1.1494-1511