
Abstract
The cel12B gene was cloned, optimized through directed evolution using error-prone polymerase chain reaction, and then expressed in the Escherichia coli BL21 (DE3) host strain. Five mutants promoting the enzyme activities were selected. The specific activity of the best-evolved Cel16 (L20R, D37V, I108T) was improved approximately 3-fold compared to the parental enzyme. The residual enzyme activity of Cel16 retained 90% of the original when incubated at 90 °C for 2 h, which was similar to the thermostability of the wild type. In addition, the best mutant Cel16, which had two prominent mutant sites, L20R and I108T, was able to increase the cavity polarity because the side chains of arginine and threonine could form hydrogen bonds with the substrate, shrinking the enzyme cavity to some extent and therefore enhancing the enzyme activity.
Download PDF
Full Article
Directed Evolution of a Hyperthermophilic Endoglucanase Cel12B from Thermotoga maritima
Hao Shi,a,b,c Lili Wang,a,b Xun Li,a,b Liangliang Wang,a,b Yu Zhang,a,b Xiangqian Li,c,* and Fei Wang a,b,*
The cel12B gene was cloned, optimized through directed evolution using error-prone polymerase chain reaction, and then expressed in the Escherichia coli BL21 (DE3) host strain. Five mutants promoting the enzyme activities were selected. The specific activity of the best-evolved Cel16 (L20R, D37V, I108T) was improved approximately 3-fold compared to the parental enzyme. The residual enzyme activity of Cel16 retained 90% of the original when incubated at 90 °C for 2 h, which was similar to the thermostability of the wild type. In addition, the best mutant Cel16, which had two prominent mutant sites, L20R and I108T, was able to increase the cavity polarity because the side chains of arginine and threonine could form hydrogen bonds with the substrate, shrinking the enzyme cavity to some extent and therefore enhancing the enzyme activity.
Keywords: Endoglucanase; Error-prone PCR; Directed evolution; Thermotoga maritima
Contact information: a: Department of Chemical Engineering of Forest Product, Nanjing Forestry University, 210037, China; b: Jiangsu Key Lab of Biomass-Based Green Fuel and Chemicals, 210037, China; c: Department of Life Science and Chemistry, Huaiyin Institute of Technology, Huaian 223003, China; *Corresponding authors: hgwf@njfu.edu.cn; lixq2002@126.com
INTRODUCTION
Cellulose is the most abundant natural polymer and renewable carbon source in the world (Wang et al. 2005). Cellulases are broadly classified into three groups based on amino acid sequence and substrate specificity: endo-1, 4-β-glucanases (EC3.2.1.4), 1,4-β-D-cellobiohydrolases (EC3.2.1.9), and 1,4-β-glucosidase (EC3.2.1.21). Endoglucanases and cellobiohydrolases can degrade cellulose to oligosaccharides and cellobiose, and glucosidase hydrolyzes cellobiose to glucose monomers (Hreggvidsson et al. 1996). For converting cellulosic biomass into fuels, the hydrolytic activities of existing industrial cellulases are insufficient for large industrial applications (Wang et al. 2010). Because of their ever increasing demand, novel cellulases with high hydrolytic activity, specificity, and stability are being discovered from new lineages of cellulolytic organisms (Nizamudeen and Bajaj 2009).
The genus Thermotoga contains one of the most extreme thermophilic eubacteria, which is able to withstand temperatures up to 90 °C (Huber et al. 1986). This genus phylogenetically represents the deepest branch off eubacteria (Achenbach-Richter et al. 1987). So far, Thermotoga maritima has been described as a type of strain which was isolated from geothermally heated marine sediment from the Azores (Huber et al. 1986). A similar organism Fervidobacterium nodosum, isolated from a New Zealand hot spring, is also a thermophilic, glycolytic anaerobic eubacterium (Zhaxybayeva et al. 2009). The investigation into the structure-function relationship of endo-1, 4-β-glucanases (EC3.2.1.4) not only can help us to understand their natural role, but also can facilitate its applications in engineering enzymes for higher specificity or converting them into the synthetic catalyst. Indeed, E. coli endo-1,4-β-glucanases have already served as a test bed for the development of technologies for protein function with directed evolution (Salleh et al. 2006). Recently, directed evolution has been widely used to improve enzymes, particularly for the industrial biocatalytic processes (Dalby 2011).
Directed evolution can be achieved without sufficient insight into the structure-function relationships. Prior to the error-prone polymerase chain reaction (epPCR), different mutagenic conditions were tested to assess which mutation would produce the best gene library. Directed evolution is able to obtain random genetic libraries and screen mutants that possess specific characteristics (Farinas et al. 2001). Random mutagenesis, one of the directed evolution methods, is regarded as a valuable protein modification toolbox with applications ranging from protein engineering to directed protein evolution studies (Rasila et al. 2009). For example, one mutant (N321T), generated by the epPCR technique and expressed in Saccharyomyces cerevisiae,was found to have an optimal pH of 5.4 with a pH shift of 0.6 (Wang et al. 2005). The examples of directed evolution for endoglucanases have been reported, and are identified by facilitated screening on solid plates containing carboxymethyl cellulose (CMC) followed by Congo Red staining (Zhang et al. 2006).
In the present study, we attempted to increase the specific activity of T. maritima in endoglucanase Cel12B in E. coli by the random mutagenesis of the gene using epPCR followed by an activity screening with plate and enzyme assays.
EXPERIMENTAL
Materials
E. coli Top10 (Novagen, Billerica, USA) was used for propagation of plasmids, and E. coli BL21 (DE3) (Novagen) was used as the host for expression. Ex-taq DNA polymerase, rTaq DNA polymerase, restriction enzymes, and dNTP were purchased from Takara (Dalian, China). Carboxymethyl cellulose was purchased from Sigma Chemicals (St. Louis, USA). Primers were synthesized by Sangon Biotech (Shanghai, China).
Methods
Growth conditions
Both the E. coli strains above were cultivated at 37 °C in Luria-Bertani (LB) medium (1% tryptone, 0.5% yeast extract, 0.5% sodium chloride, pH 7.0) supplemented with ampicillin (100 g mL-1). The expression of the recombinant enzymes in the 200 mL culture was induced by supplementing with 80 L of 1 M isopropyl -D-1-thiogalactopyranoside (IPTG) when it reached an optical density at 600 nm of approximately 0.6-0.8. The culture was incubated at 37 °C with shaking at 200 rpm for 5 h.
PCR reaction
The DNA analysis program SignalP4.0 was used to analyze the signal peptide sequence of Cel12B (GenBank NO. Z69341). Then, primers were designed to remove the signal peptide. The two primers, P1 (5′-GGAATTCCATATGAGGTGGGCAGTTCT-TCTGA-3′) and P2 (5′-CCGCTCGAGTTATTACTCGAGTTTTACACCTTCGACAG-AGAAGTC-3′), were designed with NdeI and XhoI sites, respectively. PCR was performed as follows: 94 °C for 5 min; 30 cycles of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 50 s; 72 °C for 10 min.
Random mutagenesis by error-prone PCR
We performed three rounds of mutagenesis followed by screening for increased activity of endoglucanase Cel12B using the method described by Tao et al. (2005), with dNTP concentrations of 0.2 mM dATP, 0.2 mM dGTP, 1 mM dCTP, 1 mM dTTP, 3-7 mM MgCl2, 0.1-0.3 mM MnCl2, 0.1 U rTaq DNA polymerase, and 0.2 nM DNA template (cel12B gene fragment). The primers were the same as those mentioned above. The PCR product was verified on an agarose gel and purified with gel extraction using quick DNA purification kit (Qiagen, USA), digested by NdeI/Xhol followed by purification and ligation to the pET-20b vector at the corresponding sites, and then transformed into E. coli BL21 (DE3) competent cells.
Mutant library screening
Transformants were selected by inoculating them on an LB plate containing CMC (0.1%), IPTG (isopropyl-L-D-thiogalactopyranoside, 0.1 mM), and ampicillin (100 ug/mL). The culture was incubated overnight at 37 °C and further assayed by Congo-red staining of undigested celluloses. The growth plates were exposed to 10 mM Tris-EDTA buffer (pH 8.0) to wash the cells, followed by Congo-red staining (0.1% Congo-red). After staining for 30 min at 70 °C, plates were placed at 50 °C for 30 min and washed with 1 M sodium chloride. The transformants creating a larger transparent circle than the wild type were selected as the target strains for further enzyme activity studies. Finally, mutants with the improved thermostability were grown and the corresponding plasmids were extracted and sent for DNA sequencing.
Enzyme production and purification
Verified recombinant plasmids harboring the cel12b gene were transformed into E. coli BL21 (DE3) competent cells. The transformed cells were grown in 50 mL of LB medium containing ampicillin (100 ug/mL) at 37 °C until the OD600 was in the range of 0.6 to 0.8 and were further induced by IPTG (0.4 mM), followed by 5 h of incubation. The cells were harvested by centrifugation (15000 xg for 10 min), and the resulting pellets were resuspended in Tris-HCl buffer (20 mM, pH 8.0) and lysed by sonication. The solution was heated to 70 °C for 30 min to denature the unwanted proteins. After centrifugation, the supernatant containing the target proteins was further purified using a His-Bind purification Kit (Novegen), and SDS-PAGE (12%) for visulazition of bands using protein makers (15-170 kDa, MBI Fermentas, Canada) was carried out to verify the purity of the target proteins.
Enzyme assays
A reaction mixture containing 1.0% (w/v) CMC and 0.1 mL of diluted enzyme solution was incubated in 50 mM imidazole-potassium buffer (pH 6.0) at 85 °C for 10 min. The total reaction system was 1 mL, and the reaction was stopped by adding 2 mL of 3,5-dinitrosalicylic acid (DNS) reagent, with glucose as the standard. One unit of enzyme activity was defined as the amount of enzyme that produced 1 mol of glucose equivalent per min.
Homology Modeling
The crystal structure of the thermophilic endoglucanase (PDB ID: 3ammB) was used as a template for the molecular model of endoglucanase Cel12B. Then, the aligned sequences were submitted to SWISS-MODEL (http://www.expasy.org/). Structure visualization and analysis were achieved using PyMOL 0.97.
RESULTS AND DISCUSSION
Error-prone PCR Mutagenesis of cel12B Gene, Screening Mutant Library, and Enzyme Assay
In recent years, it has become easy to carry out evolutionary processes at the lab bench (Eijsink et al. 2005). There are several technologies to generate proteins with novel functions in the lab, such as unnatural amino acid mutagenesis, non-natural cofactor engineering, and computational design (Brustad and Arnold 2011). Among these, the useful technique for directed evolution in single gene is error prone PCR, which is commonly applied to initiate a random mutation. An evolutionary strategy consisting of random mutagenesis and screening followed by recombination of a few key mutants was carried out in this study. One single round of random mutagenesis in addition to the 1500 to 3000 clones screened yielded several significant thermostable variants. However, using only one round of random mutagenesis ignores information contained in the DNA fragment. Therefore, more than two rounds of mutagenesis were carried out to construct the mutant library in this study.
The mutation frequency can be modulated using a mutagenic buffer which unbalances the nucleotide concentrations (Cadwell and Joyce 1992) and other factors such as the concentration of MgCl2, dNTP, and Taq DNA polymerase. From the agarose gel it was observed that the intensity of the bands varied with each condition applied (Fig. 1). The optimal condition in the PCR system was determined as: 7 mM MgCl2, 0.15 mM MnCl2, 0.2 mM dATP, 0.2 mM dGTP, 1 mM dCTP, 1 mM dTTP, and 5 U of Taq DNA polymerase, with a lower annealing temperature (50-54 °C) and no hot start.

Fig. 1. Agarose gel electrophoresis of epPCR product under different conditions. M, standard marker. (a) Lane l through Lane 5 (0.3 mM Mn2+; 3 mM, 4 mM, 5 mM, 6 mM, and 7 mM Mg2+, respectively) and (b) Lane 1 through Lane 5 (7 mM Mg2+; 0.1 mM, 0.15 mM, 0.2 mM, 0.25 mM, and 0.3 mM Mn2+, respectively)
Leakage of the cytoplasmic enzyme facilitates the detection of the individual colonies which express endoglucanase with increased activity using the plate-based assay as described above. The purity of the wild type enzyme and Cel16 was confirmed by SDS-PAGE (Fig. 2). Mutants with relative activity 20% higher than the wild type were selected in the enzyme assays. Subsequently, the thermostability of the selected mutants was assayed in a test tube. After two rounds of directed evolution, five highly improved T. maritima endoglucanase in terms of specific activity, thermostability, and pH stability in E. coli was obtained (Table 1). Base sequences were transformed leading to the change of amino acid sequences that mainly results in the enhancement of enzyme activity (Zhao and Arnold 1999). Most of the mutant sites were designed to be changed between the bases A and T or C and G. In our study, the enzyme activity of the mutants was 2- to 3-fold greater than the control when using CMC as substrate. The optimal enzyme, Cel16 (L20R, D37V, I108T), containing 3 amino acid changes, was approximately 3 times higher in enzyme activity than its parent clone. As shown in Fig. 3, the relative activity of Cel16 (L20R, D37V, I108T) was approximately 3 times higher (149.66) than the wild type (46.21 U/mg). In addition, the cel16 gene sequence obtained from epPCR had 97% similarity with the wild type (Amino acid sequence identity, 99%).

Fig. 2. SDS-PAGE analysis of Cel12B. Lane 1, purified enzyme of wild-type Cel12B; Lane 2, purified mutant Cel16; and Lane 3, supernatant of wild-type Cel12B after sonication
Table 1. Mutated Enzymes with Changed Sites of Nucleotide and Amino Acid using Error-prone PCR

Enzymatic Properties of Mutant Cel16
As shown in Fig. 4a, mutant Cel16 remained active within a wide range of temperatures (60-100 °C), which was comparable to its wild-type Cel12B. To determine the thermostability, Cel12B and Cel16 were incubated at 60, 65, 70, 75, 80, 85, 90, 95, and 100 °C for 2 h using 1.0% CMC in 50 mM iminazole-potassium buffer (pH 6.0) as the substrate.

Fig. 3. The specific activity of each mutant enzyme and wild type. The reaction system was 1 mL containing 1.0% (w/v) CMC and 0.1 mL of diluted enzyme solution in 50 mM imidazole-potassium buffer (pH 6.0) at 85 °C for 10 min. Values are the mean of triplicate experiments, and the variation (standard error) about the mean was below 5%.

Fig. 4. Enzymatic properties of Cel16 and wild type. (a) Effect of temperature on endoglucanase. ● Wild type, ■ Cel16. (b) Effect of temperature on thermostability of activities. ○ Wild type at 90 °C, ●Cel16 at 90 °C, □Wild type at 100 °C, ■ Cel16 at 100 °C. (c) Effect of pH on endoglucanase activities. ● Wild type, ■ Cel16. (d) Effect of pH on the stability of endoglucanase at 85 °C for 2 h. ● Wild type, ■ Cel16. The residual activity was monitored, and the maximum activity was defined as 100% (a, c) or initial activity was defined as 100% (b, d). Values shown were the mean of triplicate experiments, and the variation (standard error) about the mean was below 5%.
Figure 4b shows that both the wild type Cel12B and mutant Cel16 maintained over 90% of their endoglucanase activity after a 2 h incubation at 90 °C, indicating that it was still a thermostable enzyme. It should be mentioned that the endoglucanse activity decreased sharply after a 2 h incubation at 100 °C. The endoglucanase activity of the wild type and its mutant Cel16 was also determined in the pH range of 4.5 to 8.0 for 2 h in 50 mM imidazole-potassium buffer. As shown in Fig. 4c, both the enzymes exhibited endoglucanase activity in a broad pH range (pH 4.5 to 8.0, optimum pH 6.5). In addition, both enzymes exhibited a good stability at pH range 6.0 to 6.5 (Fig. 4d).
Metal ions played an important role in affecting the structure as well as the function of the mutant enzymes. Generally, metal ions may bind to the specific amino acid side chains of the protein, resulting in denaturing of the active sites or altering the enzyme activity by stabilizing or destabilizing its conformation (Li et al. 2011). The endoglucanase activity of Cel16 was also determined in the presence of various chemicals, to include CuSO4, CoCl2, ZnSO4, MnSO4, KCl, NaNO3, Li2SO4, MgCl2, CaCl2, and BaCl2. The results indicated that specific divalent cations, Mg2+, Ba2+, slightly activated the enzyme with a relative activity of 108% and 117%, respectively (Fig. 5). This result was similar to the wild type (data not shown).
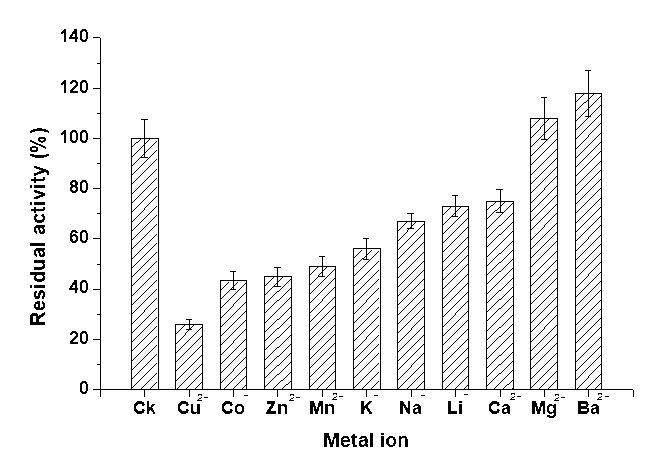
Fig. 5. Effect of metal ions on Cel16 activity. The final concentration of each cation was 1 mM in standard reaction system described in Methods above. The activity of the enzyme without adding the cation was defined as 100%. Values shown were the mean of triplicate experiments, and the variation (standard error) about the mean was below 5%.
Structure and Function Analysis of the Mutant Enzymes
Glycosyl transfer represents one of the most important biochemical reactions, which is virtually a nucleophilic substitution at the saturated carbon of the anomeric center with two hydrolytic mechanism, retention, or inversion of the anomeric configuration (Sinnott 1990), and T. maritima endoglucanase Cel12B also has a catalytic mechanism of inverting. Asp8 and Glu80 are the proton donor and nucleophile, respectively.
The three-dimensional structure of T. maritima endoglucanase Cel12B using homology modeling is shown in Fig. 6. The substrate binding region (β-sheet cavity area) of the mutant Cel16 and mutant Cel31 (data for Cel31 not shown) contain the two prominant mutant sites L20R and I108T (Fig. 6c). It can be found that L20R and I108T increase the polarity of cavity of catalytic center, make the enzyme cavity contraction to some extent, and decrease the oxo-carbenium ion balance of the distance with Glu80 residues. On the other hand, the formation of hydrogen bonds and cavity contraction lead to about 3 times enhancement in the heat resistance of the enzyme than that of the wild-type.



Fig. 6. Modeling structure of Cel12B. Segments of the secondary structure are highlighted in different colors. (a) The structure of Ce12B; (b) The catalytic sites of Cel12B (Glu80 and Asp8) are labeled red; (c) The catalytic sites of mutant Cel16 (Thr108, Arg20, Val37) are labeled black.
D37V, Y44N, and Y102H residues show different roles in the activity. The aspartic acid residue D37, located at the random coil region with a certain degree of flexibility, limited the impact on the enzyme activity. The mutant Cel31 (data not shown) with two other mutation sites (Y44N, Y102H) are located at β-sheets; thus the flexibility is limited.
CONCLUSIONS
- In this study, a library of mutant endoglucanase Cel12B from T. maritima was constructed using error-prone PCR, and five mutants promoting the enzyme activities were obtained.
- Among the mutants, the specific activity of the best-evolved Cel16 (L20R, D37V, I108T) increased approximately 3-fold compared to the wild type. In addition, the thermostability of Cel16 is nearly as same as wild type.
- Through the analysis of the three-dimensional structure of Cel12B, it is inferred that L20R and I108T have an important role in improving the enzyme activity.
ACKNOWLEDGMENTS
This work was financially supported by the National Natural Science Foundation of China (No. 31370572), the Huai’an Science and Technology Bureau (SN1143), and the Doctorate Fellowship Foundation of Nanjing Forestry University, as well as the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
REFERENCES CITED
Achenbach-Richter, L., Gupta, R., Stetter, K. O., and Woese, C. R. (1987). “Were the original eubacteria thermophiles?,” Syst. Appl. Microbiol. 9(1-2 ), 34-39.
Brustad, E. M., and Arnold, F. H. (2011). “Optimizing non-natural protein function with directed evolution,” Curr. Opin. Chem. Biol. 15(2), 201-210.
Cadwell, R. C., and Joyce, G. F. (1992). “Randomization of genes by PCR mutagenesis,” PCR Meth. Appl. 2(1), 28-33.
Dalby, P. A. (2011). “Strategy and success for the directed evolution of enzymes,” Curr. Opin. Struc. Biol. 21(4), 473-480.
Eijsink, V. G. H., Gaseidnes, S., Borchert, T. V., and van den Burg, B. (2005). “Directed evolution of enzyme stability,” Biomol. Eng. 22(1-3), 21-30.
Farinas, E. T., Bulter, T., and Arnold, F. H. (2001). “Directed enzyme evolution,” Curr. Opin. Biotech. 12(6), 545-551.
Hreggvidsson, G. O., Kaiste, E., Holst, O., Eggertsson, G., Palsdottir, A., and Kristjansson, J. K. (1996). “An extremely thermostable cellulase from the thermophilic eubacterium Rhodothermus marinus,” Appl. Enviro. Microb. 62(8), 3047-3049.
Huber, R., Langworthy, T. A., Konig, H., Thomm, M., Woese, C. R., Sleytr, U. B., and Stetter, K. O. (1986). “Thermotoga maritima sp. nov represents a new genus of unique extreme thermophilic eubacterial growing up 90 °C,” Arch. Microbiol. 144(4), 324-333.
Li, Z., Li, X., Wang, Y., Wang, Y., Wang, F., and Jiang, J. (2011). “Expression and characterization of recombinant Rhizopus oryzaelipase for enzymatic biodiesel production,” Bioresource Technol. 102(20), 9810-9813.
Nizamudeen, S., and Bajaj, B. K. (2009). “A novel thermo-alkalitolerant endoglucanase production using cost-effective agricultural residues as substrates by a newly isolated Bacillus sp NZ,” Food Technol. Biotech. 47(4), 435-440.
Rasila, T. S., Pajunen, M. I., and Savilahti, H. (2009). “Critical evaluation of random mutagenesis by error-prone polymerase chain reaction protocols, Escherichia coli mutator strain, and hydroxylamine treatment,” Anal. Biochem. 388(1), 71-80.
Salleh, H. M., Mullegger, J., Reid, S. P., Chan, W. Y., Hwang, J. Y., Warren, R. A. J., and Withers, S. G. (2006). “Cloning and characterization of Thermotoga maritima beta-glucuronidase,” Carbohyd. Res. 341(1), 49-59.
Sinnott, M. L. (1990). “Catalytic mechanisms of enzymatic glycosyl transfer,” Chem. Rev. 90(7), 1171-1202.
Tao, L., Jackson, R. E., and Cheng, Q. (2005). “Directed evolution of copy number of a broad host range plasmid for metabolic engineering,” Metab. Eng. 7(1), 10-17.
Wang, T., Liu, X., Yu, Q., Zhang, X., Qu, Y., and Gao, P. (2005). “Directed evolution for engineering pH profile of endoglucanase III from Trichoderma reesei,” Biomol. Eng. 22(1-3), 89-94.
Wang, Y., Wang, X., Tang, R., Yu, S., Zheng, B., and Feng, Y. (2010). “A novel thermostable cellulase from Fervidobacterium nodosum,” J. Mol. Catal. B 66(3-4), 294-301.
Zhang, Y. H. P., Himmel, M. E., and Mielenz, J. R. (2006). “Outlook for cellulase improvement: Screening and selection strategies,” Biotechnol. Adv. 24(5), 452-481.
Zhao, H., and Arnold, F. H. (1999). “Directed evolution converts subtilisin E into a functional equivalent of thermitase,” Protein Eng. 12(1), 47-53.
Zhaxybayeva, O., Swithers, K. S., Lapierre, P., Fournier, G. P., Bickhart, D. M., DeBoy, R. T., Nelson, K. E., Nesbo, C. L., Doolittle, W. F., Gogarten, J. P., and Noll, K. M. (2009). “On the chimeric nature, thermophilic origin, and phylogenetic placement of the Thermotogales,” P. Natl. Acad. Sci. USA. 106(14), 5865-5870.
Article submitted: January 13, 2014; Peer review completed: March 17, 2014; Revised version received: April 1, 2014; Accepted: March 11, 2014; Published: April 29, 2014.