
Abstract
Porous material was prepared by freeze drying of oxidized cellulose microfibrils. Oxidized microfibrillated cellulose was obtained by TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl)-mediated oxidation of bleached bagasse pulp and lapping process, and the influence of cellulose microfibril concentration on the material properties was studied from oxidized cellulose microfibrils within a range of solids concentrations of aqueous suspension. The microscopic morphology, pressure intensity, specific surface area, and pore size were analyzed by scanning electron microscopy (SEM), universal material testing machine, fully automatic specific surface area, and porosity analyzer (BET). The surface elemental composition of the materials was analyzed by X-ray photoelectron spectroscopy (XPS). Results showed that the strength of the porous materials increased with increasing concentration of oxidized cellulose microfibrils, along with decreases of porosity decreases, and increases of specific surface area and pore volume. A higher mass fraction resulted in a smaller pore size of the porous scaffold material and more structure of the material. However, when the concentration reached a certain value, some fiber flocculation occurred.
Download PDF
Full Article
Effects of the Solids Content of TEMPO-oxidized Cellulose Prior to Freeze-drying on the Properties of the Dried Materials
Chen Liang,a,b,* Tian Xu,a,b Xiao Wang,a,b Xin You,a,b Shuangquan Yao,a,b and Chengrong Qin a,b
Porous material was prepared by freeze drying of oxidized cellulose microfibrils. Oxidized microfibrillated cellulose was obtained by TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl)-mediated oxidation of bleached bagasse pulp and lapping process, and the influence of cellulose microfibril concentration on the material properties was studied from oxidized cellulose microfibrils within a range of solids concentrations of aqueous suspension. The microscopic morphology, pressure intensity, specific surface area, and pore size were analyzed by scanning electron microscopy (SEM), universal material testing machine, fully automatic specific surface area, and porosity analyzer (BET). The surface elemental composition of the materials was analyzed by X-ray photoelectron spectroscopy (XPS). Results showed that the strength of the porous materials increased with increasing concentration of oxidized cellulose microfibrils, along with decreases of porosity decreases, and increases of specific surface area and pore volume. A higher mass fraction resulted in a smaller pore size of the porous scaffold material and more structure of the material. However, when the concentration reached a certain value, some fiber flocculation occurred.
Keywords: Porous material; Microfibril; Oxidation; Mechanical properties; Specific surface area
Contact information: a: Department of Light Industrial and Food Engineering, Guangxi University, Nanning, 530004, PR China; b: Guangxi Key Laboratory of Clean Pulp & Papermaking and Pollution Control, Nanning 530004, PR China; *Corresponding author: liangchen@gxu.edu.cn
INTRODUCTION
Porous materials are advantageous due to their unique physicochemical properties such as low density, high porosity, specific surface area, specific strength, thermal insulation, light weight, sound insulation, high permeability, and assembly. Porous materials have broad application prospects in the fields of aerospace, adsorption, catalysis, electronic devices, biomedicine, environmental protection, and ammonia storage, etc., due to their unique physicochemical properties such as low density, high porosity, specific surface area, specific strength, thermal insulation, light weight, sound insulation, high permeability, and assembly (Vinu et al. 2006). The porous materials are prepared by biological materials, and not only have good biocompatibility, biodegradability, nontoxic character, and good cell affinity, but also have large specific surface area, high porosity, and adequate mechanical strength (Lin et al. 2018). Therefore, biomass porous materials have attracted progressively more research interest (Stein et al. 2009).
Cellulose is an important biomass resource, accounting for 59% of the content of sugar cane (Yao et al. 2017; Nie et al. 2018). Traditional sugarcane cellulose is mainly used in the paper pulping industry. Many studies have used cellulose for the preparation of functional materials, such as cellulose-based adsorbent functional materials, cellulose microfibril conductive composite materials, and cellulose packaging materials (Wegner and Jones 2006; Yao et al. 2015). Cellulose microfibril material has large specific surface area, mechanical strength, elastic modulus, tensile strength, and good thermal stability (Saito et al. 2006). The TEMPO, NaBr, and NaClO system can selectively oxidize the primary hydroxyl groups of cellulose in an aqueous solution. The oxidation of cellulose occurs on the surface of micro-fibrils, and some groups can be introduced such as carboxyl groups, aldehyde groups, and the reaction takes place without changing the fiber morphology and crystallinity (Fraschini et al. 2017). Oxidized cellulose by subsequent mild homogeneous treatment can produce nanometer-scale fibrils (Saito and Isogai 2004). The advantages of TEMPO oxidation are mild reaction conditions, simple operation, low cost, and little pollution. The nanometer fiber preparation process of low energy consumption has high yield. The resulting nanofibers have a large aspect ratio, stable dispersion in water, and are not agglomerated (Chinga-Carrasco et al. 2011).
Biological composites have been prepared with TEMPO oxidized cellulose and Cd2+ (Tang et al. 2014). Jun et al. (2017) found that TEMPO oxidized bacterial cellulose nanofibers adhere to the skin surface while maintaining nano-fibrous structures, providing inherent functions of bacterial cellulose, such as high tensile strength, high water-holding capacity, and blockage of harmful substances. Thm et al. (2018) integrated TEMPO-oxidized cellulose nanofiber at different concentrations with chitosan (CS) and created a thermosensitive injectable hydrogel intended for biomedical applications.
This paper focused on the preparation of porous scaffolds by oxidized cellulose microfibrils, including the bonding mechanism. The bleached bagasse pulp was subjected to TEMPO oxidation treatment and then freeze-dried (Sbiai et al. 2011). During the oxidation process, the hydroxyl group at the C6 position on the cellulose was selectively oxidized to the carboxyl group. The purpose of this experiment was to explore the effect of oxidized cellulose microfibril concentration prior to freeze-drying on the preparation process, strength, specific surface area, and porosity of the porous materials during the freeze-drying process.
EXPERIMENTAL
Materials
Bleached bagasse pulp was provided by a local sugar refinery (Guangxi, China). The pulp Kappa number was 14.2 and the brightness was 31.9% (%ISO). A 0.5 M sodium hydroxide solution was prepared for controlling the pH of the solution. TEMPO was purchased from Aladdin (Shanghai, China). All assay reagents were obtained from Sigma (St. Louis, MO, USA). All other chemicals employed in this work were purchased from Kawahigashi Chemical Co., Ltd. (Chong Qing, China) and were analytical grade.
Preparation of Oxidized Microfibrillated Cellulose
The TEMPO oxidation of the pulp (Isogai et al. 2011) was performed in a 1 L plastic barrel, and the pulp was adjusted to 1% (w/w) with cooled deionized water. Stirring was performed at 500 rpm, and the temperature was 10 °C. The TEMPO, NaClO, and NaBr concentrations were 0.015% (w/w), 5.2% (w/w), and 0.1% (w/w), respectively. The NaClO solution was added last. The pH of the reaction system was maintained in the range of 9.8 to 10.0 with 0.5 M NaOH. The TEMPO oxidation reaction was complete when the system was stabilized at a certain pH. The oxidized pulp was placed at 4 °C.
Porous Material Preparation Process
The oxidized nanometer cellulose was formulated at a concentration of 1%, 1.5%, 2%, and 2.3%. After freezing with a liquid nitrogen for a certain period of time (Guo et al. 2005), it was frozen into ice cubes and placed in a freeze dryer until the sample was lyophilized.
Carboxyl Content Determination
The carboxyl content (Zemljic et al. 2008) of the oxidized microfibrillated cellulose was determined by headspace gas chromatography (Chai et al. 2001). The pulp was treated with 0.1 M hydrochloric acid at room temperature for 1h. The concentration of pulp was 1.2%, and it was thoroughly washed with deionized water until it was completely free of Cl–. Next, 0.03 g of air-dried pulp samples was placed in a test bottle. After adding 4.0 mL of solution containing 0.0025 M NaHCO3 and 0.10 M NaCl, the test bottle was immediately closed and shaken so that the pulp fibers were sufficiently dispersed. The test bottle was placed into an automatic headspace sampler.
Compressive Strength Determination
An Instron-5565 universal material testing machine (Boston, MA, USA) with 2KN sensor and the accompanying Bluehill software were used (Instron). The dry mechanical properties of scaffold material were tested at a compression rate of 1 mm/min; the humidity conditions were 23 °C ± 2 °C and 50% ± 5%. To evaluate the measurement error, each sample was measured five times, and the relative standard deviation was calculated.
Morphology Analysis
Before the SEM measurements, the freeze-dried samples were cut into flakes. The scanning electron microscopy (SEM, 3400-I, Hitachi, Tokyo, Japan) images were used to analyze the morphology of the freeze-dried scaffolded material. The sample was pasted on the sample table with conductive adhesive. The internal appearance was observed after spraying gold, and the acceleration voltage for the measurement was 10 kV.
Porosity and Specific Surface Area
The porosity of the material was determined by the gravimetric method (Liu et al. 2017b). Using the measured density of individual materials (da) and wood fibers (dn; approximately 1.5 g/cm3), the gas density (P) obtained from simple mixing rules was almost negligible, as shown in Eq. 1:
P (%) = (1-da/dn) × 100 (1)
A nitrogen adsorption-desorption test was used to characterize the pore size distribution and specific surface area of the analytical materials (Xu et al. 2017). The specific surface area and pore structure of the composites were analyzed by a low temperature adsorption-desorption test. Prior to testing, the sample was degassed at 100 °C for 24 h, and the N2 adsorption-desorption isotherm of the sample was measured at liquid nitrogen temperature (-196 °C) (Liu et al. 2017a). The specific surface area of the sample was calculated (Groen et al. 2003; Tang et al. 2014). The pore size distribution of the sample was estimated by the HK method. The total pore volume of the sample and average pore size was estimated by N2 adsorption at a relative pressure (P/P0) of 0.99.
XPS Analysis of Porous Materials
The surface functional groups of the porous materials were determined by photoelectron spectroscopy (XPS, Kratos Axis Ultra DLD, Kyoto, Japan). XPS was used to confirm the chemical composition of the polymeric surfaces (Chen and Tanaka 1998). The data for at least two samples of each type of polymer film were acquired with a Riber LAS-3000 system (Tokyo, Japan) using Mg·Kα X-rays (1253.6 eV, non-monochromated) from a Mg-Al dual source. The pass energy was 20 eV and had a spot size of 1 mm. The kinetic energy of the resultant photoelectrons was analyzed. The base pressure of the main analytical chamber was 2×10-9 Torr. Survey scans from 0 eV to 1200 eV were acquired with a step size of 1.0 eV and a dwell time of 0.1 s. Narrow scans of individual photo peaks were acquired with a step size of 0.1 eV and a dwell time of 0.2 s (Huang and Xu 2014).
RESULTS AND DISCUSSION
Analysis of the Properties of Oxidized Microfibrillated Cellulose
Particle size
The length and width distribution of the fibrils that were measured using a fiber analyzer, as shown in Fig. 1.
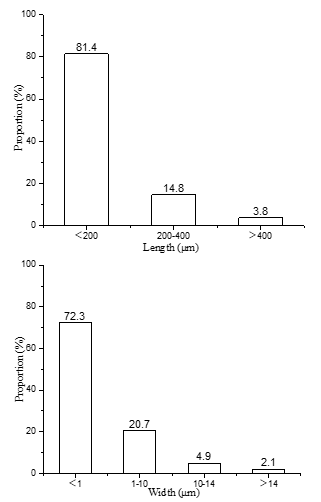
Fig. 1. Length and width distribution of the cellulose microfibrils
The results showed that 81.4% of the filament length distribution was less than 200 μm, and 14.8% of the filament length was distributed in the range 200 μm to 400 μm. There was 72.3% of the filament width distribution that was less than 1 μm. There was 20.7% of the filament width distribution between approximately 1 μm to 10 μm. In general, the sugarcane fibrils had a length of approximately 1.0 mm to 2.0 mm and a width of approximately 14 μm to 28 μm.
The length of the cellulose microfibrils was mainly distributed between less than 200 μm, and the width was mainly distributed between less than 1 μm, indicating that the oxidation process not only introduces new radicals. In the group, the refining process could reduce the size of the fibers within a certain range.
Carboxyl content
The carboxyl content of the microfibrillated cellulose was measured by a headspace gas phase method (Fujisawa et al. 2011). The carboxyl content of the different types of oxidized cellulose microfibrils is shown in Table 1. It can be observed from the Fig. 1 that the oxidation process increased the carboxyl content from 12.13 mmol.100g-1 to 81.9 mmol.100g-1, and the mechanical treatment increased the carboxyl content to 83.0 mmol.100g-1.
The mechanical treatment reduced the particle size of the fiber and increased the contact area. The main reason for the increase in the carboxyl content may be due to the exposure of more groups.
Table 1. Carboxyl Content of Different Types of Oxidized Microfibrillated Cellulose

Effect of Oxidized Microfibrillated Cellulose Concentration on Mechanical Properties of Porous Materials
The compressive strength of the material was calculated from the first linear region in the compressive stress-strain curve using the strength of the test material of the universal material testing machine, and the compressive strength at the end of the first linear region was taken as the final compressive strength of the material (Joseleau et al. 2012).
Figure 2 shows that when the concentration increased, the compressive strength and compressive modulus of the material also gradually increased. The compressive strength rapidly increased from 1 % to 2 %, due to the increase of the concentration resulting in an increase in the amount of cellulose microfibril per unit volume, while increasing the association between functional groups of the cellulose microfibril. When the concentration was more than 2 %, the increase of the compressive strength tended to be saturated.
Due to the concentration that continued to increase, the number of fibers per unit volume tended to be saturated and the fibers began to flocculate.

Fig. 2. The effect of the concentration of dry pulp on the compressive strength
Effect of Oxidized Microfibrillated Cellulose Concentration on the Porosity and Specific Surface Area of Porous Materials
The porosity of the material was measured using the gravimetric method, and the specific surface area and pore size of the material were measured by BET. Figure 3 shows that the pore size distribution of porous materials prepared with different concentrations of cellulose microfibril was very wide, and most of them were in the range of 2 nm to 120 nm, indicating that there was a notable amount of mesopores in the porous materials (Chu et al. 2016).

Fig. 3. Pore size distribution of porous materials
The pore size distribution of the porous material had two peaks at 3 nm and 32 nm, and the porosity of the porous material prepared by different concentrations of microfibrillated cellulose was somewhat different. The N2 adsorption and desorption isotherm curve was H3 (Groen et al. 2003), and the peak at 3 nm was a false peak, indicating that the pore channels in the microfibrillated cellulose porous material were connected. The peaks at 1%, 1.5%, 2%, and 2.3% content of microfibrillated cellulose were more pronounced at 32 nm, which indicated that the pores of the porous material were regular, the pore distribution was relatively uniform, and the majority were mesopores.
The oxidized microfibrillated cellulose porous materials were dehydrated at 100 °C for 24 h (Deng et al. 2017). The N2 adsorption and desorption isotherm curve of porous materials prepared with different concentrations of microfibrillated cellulose is shown in Fig. 4. It is apparent from Fig. 4 that the P/P0 in the relatively low initial stage, in accordance with the classification of International Union of Pure and Applied Chemistry (IUPAC), the isothermal adsorption curve of the N2 adsorption and desorption isotherm of the porous material prepared by freeze-drying was the same as that of the type II pressure (Stein et al. 2009; Bang et al. 2017; Beltrame et al. 2018). At relatively low but increasing pressure the porous material underwent a transition from monolayer to multilayer adsorption, and large pores existed in the porous material. When the relative pressure increased to a certain extent, the isothermal adsorption and desorption curve of N2 formed a hysteresis loop, which indicated that the sample with a rich mesoporous structure were consistent with the literature.

Fig. 4. Adsorption / desorption curve of N2 of porous materials

Fig. 5. Effect of percent drying percent on porosity of porous materials
Figure 5 shows that the percentages of oxidized microfibrillated cellulose were 1%, and the porosity was the highest at approximately 99%. When the percentage of the microfibrillated cellulose concentration increased, the porosity declined. When the pulp concentration percentage content increased to 2.3 %, the porosity dropped to approximately 98%. As the content of oxidized microfibrillated cellulose increased, the number of molecules per unit volume increased, which increased the density of the molecular network, resulting in the intertwined multiple molecules that were entangled with each other. At the formation of the porous material, the higher concentration initiated intermolecular association, making it easier to form a small, uniform condensed phase, resulting in a reduction of the porosity of the porous material. It is apparent from Table 2 that the specific surface area and pore volume also increased with the increase of the concentration of oxidized microfibrillated cellulose in the porous material. The specific surface area of porous materials and the pore volume showed increasing trends, and the average pore size decreased gradually.
Table 2. Pore Structure of Oxidized Microfibrillated Cellulose Porous Materials

The Effect of Oxidized Microfibrillated Cellulose Concentration on the Surface Morphology of Porous Materials
Different concentrations of oxidized microfibrillated cellulose porous scaffolded material were analyzed by SEM (Fig. 6). The materials were observed at 300x magnification. When the mass fraction was 1%, the SEM showed that the surface of the scaffold material was not smooth, with a large number of collapsed pores, and the distribution of surface pore sizes was broad. When the mass fraction was 1.5%, the surface of the scaffold material tended to be flat, with only a small part collapsed, and the gap between the apertures gradually decreased. When the microfibrillated cellulose mass fraction (CMF) was increased from 2% to 2.3% the majority of pores were uniform, and the particle size distribution was relatively narrow. However, when the mass fraction was 2.3%, the phenomenon of slight fiber flocculation occurred, even though the uniform pore size was flat. The higher mass fraction in aqueous suspension resulted in a smaller pore size in porous scaffolds and more structure regularity. However, the flocculation of the fibers occurred when the concentration reached a certain value.
Porous Material Surface Bonding Method Analysis
The surface characteristics of microfibrillated cellulose porous materials were analyzed by XPS (Chen and Tanaka 1998; Chen et al. 2000) (Table 3 and Fig. 7). Cellulose can be divided into four types of carbon atoms based on the form of binding (Chen and Tanaka 1998): C1 (C-C/C-H), C2 (C-OH), C3 (O-C-O/C=O), and C4 (O=C-O). Before oxidation, the starting material contains only C1, C2, C3, and C2, which indicates that the hydroxyl content was much larger than other groups (Yang et al. 2010). The oxidation process produced a large amount of carboxyl groups.

Fig. 6. Different concentrations of oxidized pulp prepared porous material: concentration of 1% oxidized pulp porous material (a), concentration of 1.5% oxidized pulp porous material (b), concentration of 2% oxidized pulp porous material (c), concentration of 2.3% oxidized pulp porous material (d).
With the increase of the concentration of porous materials, the percentages of hydroxyl and carboxyl groups associated with C2 and C4 on the surface were gradually reduced. This is likely due to the lower content of radicals when the concentration was lower. The polymerization of the molecules decreases, resulting in a lower strength of the material. As the concentration increased, the content of hydroxyl and carboxyl groups increased and the surface area of contact increased, which formed more hydrogen bonds. The increase of hydrogen bonds was the main reason for the increase of the strength of porous materials.
Table 3. Porous Material Surface XPS Analysis C Ratio


Fig. 7. XPS spectra of different concentrations oxidized cellulose: bleached bagasse pulp concentration of 2% (a), oxidized pulp at a concentration of 1% (b), oxidized pulp at a concentration of 2% (c)
CONCLUSIONS
- TEMPO system can be used to oxidize cellulose, and the carboxyl group was 82.9 mmol.100g-1. The length of the cellulose microfibrils was mainly less than 200 μm, and the width was mainly less than 1 μm. When the compressive strength of porous material was gradually increased, and the porosity gradually decreased, the specific surface area and pore volume also increased, but the pore size decreased. The mesopore content generally do not change with concentration.
- When the concentration was 2%, the scaffold material presented a porous network structure, the surface was smooth, the pore size was uniform, the pore distribution was regular. When the pore size was smaller, there was a more regular structure. When the mass fraction was more than 2%, the pore distribution of the scaffold material was still regular but slight fiber flocculation began to appear. With the increase of the oxidized pulp concentration, the compressive strength of the material also gradually increased, and the porous material formed a large number of hydrogen bonds to increase the strength. As the concentration of CMF was increased, the specific surface area and pore volume of the prepared porous material increased gradually, and the pore size decreased.
ACKNOWLEDGMENTS
This project was sponsored by the National Natural Science Foundation of China (Grant No. 21466004 and 31760192), the Guangxi Natural Science Foundation of China (Grant No. 2016GXNSFBA380234), and the Guangxi Special Funds for Academic and Technical Leaders.
REFERENCES CITED
Bang, J., Lee, H., An, K., and Kim, B. (2017). “A study on optimal pore development of modified commercial activated carbons for electrode materials of supercapacitors,” Applied Surface Science 415, 61-66. DOI: 10.1016/j.apsusc.2017.01.007
Beltrame, K., Cazetta, A., de Souza, P., Spessato, L., Silva, T., and Almeida, V. (2018). “Adsorption of caffeine on mesoporous activated carbon fibers prepared from pineapple plant leaves,” Ecotoxicology and Environmental Safety 147, 64-71. DOI: 10.1016/j.ecoenv.2017.08.034
Chai, X., Luo, Q., and Zhu, J. (2001). “Analysis of nonvolatile species in a complex matrix by headspace gas chromatography,” Journal of Chromatography 909(2), 249-257. DOI: 10.1016/S0021-9673(00)01085-2
Chen, S., and Tanaka, H. (1998). “Surface analysis of paper containing polymer additives by X-ray photoelectron spectroscopy I: Application to paper containing dry strength additives,” Journal of Wood Science 44(4), 303-309. DOI: 10.1007/BF00581311
Chen, S., Wu, Z., and Tanaka, H. (2000). “Surface analysis of paper containing polymer additives by X-ray photoelectron spectroscopy II. Paper containing N-chloropolyacrylamide,” Journal of the Japan Wood Research Society 46(5), 456-461.
Chinga-Carrasco, G., Yu, Y., and Diserud, O. (2011). “Quantitative electron microscopy of cellulose nanofibril structures from Eucalyptus and Pinus radiata kraft pulp fibers,” Microscopy and Microanalysis 17(4), 563-571. DOI: 10.1017/S1431927611000444
Chu, H., Wang, Z., and Liu, Y. (2016). “Application of modified bentonite granulated electrodes for advanced treatment of pulp and paper mill wastewater in three-dimensional electrode system,” Journal of Environmental Chemical Engineering 4(2), 1810-1817. DOI: 10.1016/j.jece.2016.02.017
Deng, L., Lu, B., Li, J., Lv, G., Du, S., Shi, J, and Yang, Y. (2017). “Effect of pore structure and oxygen-containing groups on adsorption of dibenzothiophene over activated carbon,” Fuel200, 54-61. DOI: 10.1016/j.fuel.2017.03.018
Fraschini, C., Chauve, G., and Bouchard, J. (2017). “TEMPO-mediated surface oxidation of cellulose nanocrystals (CNCs),” Cellulose 24, 2775-2790. DOI: 10.1007/s10570-017-1319-5
Fujisawa, S., Okita, Y., Fukuzumi, H., Saito, T., and Isogai, A. (2011). “Preparation and characterization of TEMPO-oxidized cellulose nanofibril films with free carboxyl groups,” Carbohydrate Polymers 84(1), 579-583. DOI: 10.1016/j.carbpol.2010.12.029
Groen, J., Peffer, L., and Perez-Ramirez, J. (2003). “Pore size determination in modified micro- and mesoporous materials. Pitfalls and limitations in gas adsorption data analysis,” Microporous and Mesoporous Materials 60(1-3), 1-17. DOI: 10.1016/S1387-1811(03)00339-1
Guo, X., Xiao, P., Liu, J., and Shen, Z. (2005). “Fabrication of nanostructured hydroxyapatite via hydrothermal synthesis and spark plasma sintering,” Journal of the American Ceramic Society 88(4), 1026-1029. DOI: 10.1111/j.1551-2916.2005.00198.x
Isogai, A., Saito, T., and Fukuzumi, H. (2011). “TEMPO-oxidized cellulose nanofibers,” Nanoscale 3(1), 71-85. DOI: 10.1039/c0nr00583e
Joseleau, J., Chevalier-Billosta, V, and Ruel, K. (2012). “Interaction between microfibrillar cellulose fines and fibers: Influence on pulp qualities and paper sheet properties,” Cellulose19(3), 769-777. DOI: 10.1007/s10570-012-9693-5
Jun, S., Lee, S., Kim, S., Park, S., Lee, C., and Kang, N. (2017). “Physical properties of TEMPO-oxidized bacterial cellulose nanofibers on the skin surface,” Cellulose 24(12), 1-8. DOI: 10.1007/s10570-017-1508-2
Lin, X., Wu, Z., Zhang, C., Liu, S., and Nie, S. (2018). “Enzymatic pulping of lignocellulosic biomass,” Industrial Crops and Products 120, 16-24. DOI: 10.1016/j.indcrop.2018.04.033
Liu, Y., Chen, Y., Tian, L., and Hu, R. (2017a). “Hierarchical porous nitrogen-doped carbon materials derived from one-step carbonization of polyimide for efficient CO2 adsorption and separation,” Journal of Porous Materials 24(3), 583-589. DOI: 10.1007/s10934-016-0294-9
Liu, Y., Lu, P., Xiao, H., Heydarifard, S., and Wang, S. (2017b). “Novel aqueous spongy foams made of three-dimensionally dispersed wood-fiber: Entrapment and stabilization with NFC/MFC within capillary foams,” Cellulose 24(1), 1-11. DOI: 10.1007/s10570-016-1103-y
Nie, S., Zhang, K., Lin, X., Zhang, C., Yan, D., Liang, H., and Wang, S. (2018). “Enzymatic pretreatment for the improvement of dispersion and film properties of cellulose nanofibrils,” Carbohydrate Polymers 181, 1136-1142. DOI: 10.1016/j.carbpol.2017.11.020
Thm, N., Abueva, C., Ho, H., Lee, S., and Lee, B. (2018). “In vitro and in vivo acute response towards injectable thermosensitive chitosan/TEMPO-oxidized cellulose nanofiber hydrogel,”Carbohydrate Polymers 180, 246-255. DOI: 10.1016/j.carbpol.2017.10.032
Saito, T., and Isogai, A. (2004). “TEMPO-mediated oxidation of native cellulose. The effect of oxidation conditions on chemical and crystal structures of the water-insoluble fractions,” Biomacromolecules 5(5), 1983-1989. DOI: 10.1021/bm0497769
Saito, T., Nishiyama, Y., Putaux, J., Vignon, M., and Isogai A. (2006). “Homogeneous suspensions of individualized microfibrils from TEMPO-catalyzed oxidation of native cellulose,” Biomacromolecules 7(6), 1687-1691. DOI: 10.1021/bm060154s
Sbiai, A., Kaddami, H., Sautereau, H., Maazouz, A., and Fleury, E. (2011). “TEMPO-mediated oxidation of lignocellulosic fibers from date palm leaves,” Carbohydrate Polymers 86(4), 1445-1450. DOI: 10.1016/j.carbpol.2011.06.005
Stein, A., Wang, Z., and Fierke, M. 2009. “Functionalization of porous carbon materials with designed pore architecture,” Advanced Materials 21(3), 265-293. DOI: 10.1002/adma.200801492
Tang, A., Zhao, S., and Song, J. (2014). “Structure control and characterization of 3D porous scaffold based on cellulose-nanofibers for tissue engineering,” Chinese Journal of Materials Research 28(10), 721-729.
Vinu, A., Mori, T., and Ariga, K. (2006). “New families of mesoporous materials,” Science and Technology of Advanced Materials 7(8), 753-771. DOI: 10.1016/j.stam.2006.10.007
Wegner, T., and Jones, P. (2006). “Advancing cellulose-based nanotechnology,” Cellulose 13(2), 115-118. DOI: 10.1007/s10570-006-9056-1
Xu, Z., Zhang, D., Yuan, Z., Chen, W., Zhang, T., Tian, D., and Deng, H. (2017). “Physicochemical and adsorptive characteristics of activated carbons from waste polyester textiles utilizing MgO template method,” Environmental Science and Pollution Research 24(28), 1-11. DOI: 10.1007/s11356-017-9939-8
Yang, S., Jiang, Z., Ren, H., and Fei, B. (2010). “Determination of cellulose crystallinity of bamboo culms with X-ray diffraction spectrum,” Journal of North-East Forestry University 38, 75-77.
Yao, S., Nie, S., Yuan, Y., Wang, S., and Qin, C. (2015). “Efficient extraction of bagasse hemicelluloses and characterization of solid remainder,” Bioresource Technology 185, 21-27. DOI: 10.1016/j.biortech.2015.02.052
Yao, S., Nie, S., Zhu, H., Wang, S., Song, X., and Qin C. (2017). “Extraction of hemicellulose by hot water to reduce adsorbable organic halogen formation in chlorine dioxide bleaching of bagasse pulp,” Industrial Crops and Products 96, 178-185. DOI: 10.1016/j.indcrop.2016.11.046
Zemljic, L., Persin, Z., Stenius, P., and Kleinschek, K. (2008). “Carboxyl groups in pre-treated regenerated cellulose fibres,” Cellulose 15(5), 681-690. DOI: 10.1007/s10570-008-9216-6
Article submitted: May 4, 2018; Peer review completed: July 13, 2018; Revised version received: October 8, 2018; Accepted: October 9, 2018; Published: October 22, 2018.
DOI: 10.15376/biores.13.4.8871-8884