
Abstract
Lignin, which is the most abundant aromatic polymer in nature, was used as a green substitute for the toxic bisphenol A. In particular, the ability of epoxidized lignin to simultaneously serve as a cross-linker and rigid segment was investigated. The epoxidized lignin was preferably reacted with a monofunctional amine, which acted as a chain extender, to evaluate its performance as a cross-linker, and in the presence of poly(ethylene glycol) diglycidyl ether as a soft segment to adjust the resin properties. Different poly(ethylene glycol) diglycidyl ether/lignin stoichiometric ratios were tested, whereas the amine/epoxy equivalent ratio was fixed at 1:2. Some of the remarkable resin samples were subjected to differential scanning calorimetry analysis and compared with blank samples that did not include lignin in the composition. Moreover, the evolution over time of the molecular weight distribution of the selected compositions was analyzed by gel permeation chromatography until the solubility in tetrahydrofuran was appreciable.
Download PDF
Full Article
Epoxidized Lignin Derivatives as Bio-based Cross-linkers Used in the Preparation of Epoxy Resins
Anika Salanti,a Luca Zoia,a,* Roberto Simonutti,b and Marco Orlandi a
Lignin, which is the most abundant aromatic polymer in nature, was used as a green substitute for the toxic bisphenol A. In particular, the ability of epoxidized lignin to simultaneously serve as a cross-linker and rigid segment was investigated. The epoxidized lignin was preferably reacted with a monofunctional amine, which acted as a chain extender, to evaluate its performance as a cross-linker, and in the presence of poly(ethylene glycol) diglycidyl ether as a soft segment to adjust the resin properties. Different poly(ethylene glycol) diglycidyl ether/lignin stoichiometric ratios were tested, whereas the amine/epoxy equivalent ratio was fixed at 1:2. Some of the remarkable resin samples were subjected to differential scanning calorimetry analysis and compared with blank samples that did not include lignin in the composition. Moreover, the evolution over time of the molecular weight distribution of the selected compositions was analyzed by gel permeation chromatography until the solubility in tetrahydrofuran was appreciable.
Keywords: Lignin; Cross-linker; Epoxy resin; Differential scanning calorimetry (DSC); Gel permeation chromatography (GPC)
Contact information: a: Department of Earth and Environmental Sciences, University of Milano-Bicocca, Piazza della Scienza, 1 Milan, I-20126 Italy; b: Department of Materials Science, University of Milano-Bicocca, Via R. Cozzi, 55 Milan, I-20125 Italy; *Corresponding author: luca.zoia@unimib.it
INTRODUCTION
The main purpose of green chemistry is the design of chemical products and processes that reduce or eliminate the generation of toxins and waste. In the field of plastics chemistry, achieving these requirements essentially means avoiding toxic intermediates and by-products, as well as energy- and solvent-demanding processes, while reducing the demand of finite fossil-based resources. The development of bio-based polymers is crucial to overcome the challenges of a more sustainable and aware society. Chemicals obtained from alternative, renewable feedstocks are currently under investigation (Ng et al. 2017).
Epoxy resins are known for the versatile nature of their structure and applications. They are for the most part exploited in the paint and coating industry, due to their ability to form tough, thermo-stable and highly-resistant layers in protection to different materials. Other markets include electrical, composites, and plastic manufacturing.
The first epoxy resin was developed in 1936 by Pierre Castan. He produced a bisphenol A (BPA, 4,4’-(propane-2,2-diyl)diphenol))-based epoxy resin via reaction with epichlorohydrin, and subsequently prepared a thermoset composition by reacting the pre-polymer with phthalic anhydride (Castan 1943). The strategy for the synthesis of pre-polymers has not really changed since then, and approximately 90% of epoxy cross-linked resins are still obtained from BPA, which is a known reprotoxic and endocrine disruptor substance (Dodds and Lawson 1936; Becker et al. 2009). This is because the characteristics provided by the presence of BPA in the epoxy network are difficult to replicate since its aromatic rings confer excellent mechanical properties and thermal resistance.
Fortunately, a safer and more environmentally friendly process for the production of epichlorohydrin has been developed based on the chlorination of glycerol, which is a by-product of the biodiesel industry (Santacesaria et al. 2009). Nevertheless, extensive academic and industrial research is ongoing to find suitable alternatives to BPA (Gandini 2010; Auvergne et al. 2014; Baroncini et al. 2016). Bio-based building blocks can be attained from many natural sources, such as phenols, polyphenols, lignin, lignin-derived phenols, polyols, and vegetable oils. Bisphenol A analogues can be prepared via electrophilic aromatic condensation of vanillyl alcohol and guaiacol to obtain bisguaiacol (Hernandez et al. 2016), through the acylation of the phenolic hydroxyl group of isoeugenol with terephthaloyl chloride (Wan et al. 2015) and by O-glycidylation of gallic acid, 4-methyl cathecol, and resorcinol (Aouf et al. 2013). Lignin, as a multifunctional macromolecule bearing diverse groups prone to epoxidation, has been used to prepare highly networked resins (Koike 2012; Asada et al. 2015; Xin et al. 2016), while some of its degradation products, such as vanillin (Koike 2012), dicarboxylic acids (Aouf et al. 2012), and C2-acetals originating from the selective cleavage of the β-O-4 linkage (Kaiho et al. 2016), are employed as pre-polymers after epoxidation. Different bio-based linear and cycloaliphatic polyols, such as glycerol, sorbitol, isosorbide, and pentaerythritol, can be successfully epoxidized with epichlorohydrin and are used as components in various resin formulations (Jordan et al. 1970; Shibata and Nakai 2010; Shibata et al. 2013). Other attempts aimed at the substitution of BPA through the exploitation of polyols have involved the esterification of pentaerythritol, dipentaerythritol, and trimethylolpropane with ethyl-4-hydroxybenzoate, and subsequently undergo glycidylation to increase the mechanical properties of pre-polymers, which are otherwise poor because of the rather flexible nature of the aliphatic segment (Fourcade et al. 2013). Plant oils are widely available and are the most employed renewable resource in the polymer chemical industry as plasticizers and stabilizers. Triglycerides from vegetable oils naturally contain several functional groups, such as double bonds and hydroxyls, which can be readily epoxidized. Epoxidized linseed, soybean, and castor oils are among the most exploited oils in resin formulations (Park et al. 2004; Supanchaiyamat et al. 2012). In all cases, the mechanical and thermal properties of the resulting thermoset epoxy resins were found to greatly vary as a function of the chemical structure of the epoxy pre-polymer and its substituents, as well as the chemical structure of the curing agent. In general terms, when the pre-polymer is more flexible, the mechanical properties are poorer, and the glass transition temperature (Tg) of the epoxy resin is lower.
In accordance with the above-mentioned approaches, the purpose of the present work was the valorization of lignin as a macromonomer and structural component in epoxy resins. Lignin is the second most abundant polymer in the world, behind cellulose, and it is the ubiquitous aromatic macromolecule. Despite its widespread availability, mainly as a side-product in different processes based on lignocellulosic transformation, and copious studies aimed at its exploitation, the utilization of lignin is still hampered by the widely varying chemical and morphological structure of the macromolecule. Nevertheless, by virtue of its cross-linked structure and large amount of reactive phenolic functionalities, possible applications of lignin are being widely researched by the scientific community.
The conversion of lignin into high-value polymeric materials has been recently reviewed by Upton and Kasko (2016). Many recent studies exploiting lignin as an epoxy pre-polymer are characterized by a preliminary depolymerization step; low molecular weight lignin is obtained and then subjected to glycidylation and cured. Reducing the molecular weight is a valuable approach to increase the functionalization and miscibility with other resin components because the highly branched structure of lignin often prevents a thorough glycidylation of the phenolic groups (Ferdosian et al. 2014, 2015, 2016). An alternative method to increase the amount of epoxide groups is to subject pristine lignin to a preliminary phenolation reaction to improve the functionalization and miscibility (Simionescu et al. 1993; Zhao et al. 2001). Only a few studies have researched the utilization of non-depolymerized, non-perfunctionalized lignin in cured formulations (Hofmann and Glasser 1993; Sasaki et al. 2013). The results of these works attested to the great potential that is connected with the utilization of lignin, avoided additional degradation processes from an energetic and environmental point of view, and highlighted the necessity of improving and tuning the mechanical and thermal properties of the resulting resin for feasible industrial exploitation.
In the present work, bio-based epoxy resins were prepared by exploiting epoxidized lignin (LignEP) as both a hard segment and cross-linker. The LignEP was preferably reacted with 1-octylamine (C8NH2), which served as a chain extender, in the presence of poly(ethylene glycol) diglycidyl ether (PEGEP) as a soft segment to tune the resin properties. The preparation and chemical characterization of the LignEP and an example of a prospective utilization of lignin as a cross-linker has already been reported by the authors (Salanti et al. 2016a,b, 2017). Some remarkable resin samples were subjected to differential scanning calorimetry (DSC) analysis to compare their Tg values to that of blank samples that did not contain lignin in the formulation, while the effect of time on the molecular weight distributions of a particular formulation was analyzed by gel permeation chromatography (GPC) until the solubility in tetrahydrofuran (THF) was appreciable. Moreover, the procedure used for the epoxidation reaction was improved, which resulted in a substantial increase in the yield of the LignEP derivative.
EXPERIMENTAL
Materials
Reagents and materials
All of the reagents and solvents used in this work were purchased from Sigma-Aldrich (Saint Louis, MO, USA) and used as received. The lignin used was supplied during the project ProLignin, EraNets WoodWisdom/Bioenergy in 2012 to 2014. It was an herbaceous lignin (purity 98.2%; Mn ≈ 2700 estimated by GPC-ultraviolet (UV) analysis; total phenolic content = 3.55 mmol/g calculated by quantitative 31P nuclear magnetic resonance (NMR) analysis) obtained from a soda pulping process.
Lignin epoxidation
One gram of dry lignin (3.55 mmol of phenol moieties) was added to 20 mL of distilled water containing KOH (2.5 g, 44.6 mmol). After complete dissolution, tetrabutylammonium bromide (TBAB, catalytic, 200 mg) and epichlorohydrin (5 mL, 63.8 mmol) were added. The mixture was reacted at 70 °C for 90 min under vigorous stirring, and then slowly poured into cold, acidified distilled water (250 mL of water containing 5 mL of 99% acetic acid) to precipitate the LignEP. The LignEP was filtered on a sintered glass crucible (size 4) and washed with acidified distilled water. When almost dry, the lignin was re-dissolved in acetone (approximately 100 mL) and the soluble fraction was recollected as the filtrate. A small amount of LignEP was left on the filter as a solid residue (approximately 100 mg). The filtrate was finally desiccated by rotary evaporation and dried in vacuum. A total of 1.1 g of purified LignEP was obtained, which implied a 92% yield based on the theoretical weight percent gain.
Model aminolysis reaction
Model aminolysis reactions were performed on the LignEP (Mn ≈ 3000 estimated by GPC-UV analysis; epoxy content = 2.34 mmol/g calculated by quantitative 31P NMR analysis) in the presence of 1-octylamine (MW = 129.2 g/mol; d = 0.782 g/mL; amine equivalent weight = 64.6 g/eq; functionality: 2), either in excess or in a stoichiometric amount. In the first case, 1 mL of C8NH2 was added to 100 mg of LignEP and the mixture was left to react at 80 °C for 20 h under stirring. In the second case (stoichiometric NH2: epoxy ratio = 1:2), 18.7 µL of C8NH2 were added to 100 mg of LignEP along with 500 µL of DMF to obtain a flowing system. After 20 h at 80 °C, the products were precipitated out in cold diethyl ether and filtered.
General procedure for the preparation of the epoxy resins
In a typical experimental procedure, PEGEP (Mn ≈ 500 as declared by Sigma Aldrich; epoxy equivalent weight ≈ 250 g/eq; functionality: 2) was put into an 8-mL screw top glass vial equipped with a magnetic stirrer. A certain amount of LignEP (Mn ≈ 3000 estimated by GPC-UV analysis; epoxy content = 2.34 mmol/g calculated by quantitative 31P NMR analysis) was then added, according to the PEGEP/LignEP stoichiometry, which is reported in Table 2, along with 250 µL of THF. After dissolution, a specific amount of 1,12-diaminododecane (DAD; MW = 200.4 g/mol; amine equivalent weight = 50.1 g/eq; functionality: 4) or 1-octylamine (MW = 129.2 g/mol; d = 0.782 g/mL; amine equivalent weight = 64.6 g/eq; functionality: 2) was added to the solution, according to the total epoxy groups/total amine equivalent ratio of 2:1. The vial was then placed in an oil bath and heated to 80 °C (to favor the melting of DAD). After 20 h, the product was cured in an oven at 80 °C for an additional 20 h. For a selected sample, the effect of time on the molecular weight distribution was determined by GPC analysis; a blank reference sample without LignEP was also prepared and benzoylated in pyridine/benzoyl chloride to obtain a UV reference. Moreover, selected samples were subjected to DSC analysis.
Lignin acetylation
If required, lignin acetylation was performed overnight in a pyridine-acetic anhydride solution (1:1 v/v, 4 mL). After stripping with ethanol, toluene, and chloroform (25 mL for each solvent), the samples were dried under vacuum.
Methods
FT-IR analysis
Fourier transform infrared spectroscopy (FR-IR) analyses were performed on a Nicolet iS10 spectrometer (Thermo Fisher Scientific, Waltham, MA, USA ) equipped with a Smart iTR™ Total Attenuated Reflectance (ATR) sampling accessory (total scan 16, range 4000 cm-1 to 700 cm-1, resolution 1 cm-1).
31P-NMR analysis
The 31P NMR analysis was performed according to a previously reported procedure (Salanti et al. 2016a), and using 2-chloro-1,3,2-dioxaphospholane as a derivatizing agent.
13C NMR analysis
Broadband decoupled 13C NMR spectra were recorded on a Bruker Avance 500 MHz spectrometer (Billerica, MA, USA) at room temperature with 800-µL samples dissolved in deuterated dimethylsulfoxide (DMSO-d6). The chemical shifts were referred to as the methoxyl group signal (56 ppm). Quantitative 13C NMR spectra were acquired in the presence of epibromohydrin (10 µL, 98%) as an internal standard. A relaxation delay of 10 s was applied between the scans (90° pulse angle), and approximately 10000 scans were accumulated. A line broadening of 0.8 Hz was applied to the FIDs before FT-IR. The assignment of the predominant signals was based on data from the literature (Salanti et al. 2016a; Spectral Database of Organic Compounds SDBS 2017).
Polymer solvation test
Accurately weighed resin samples (approximately 150 mg) were put in desiccated 8-mL screw-top glass vials and soaked with THF (8 mL) at room temperature for 20 h with occasional shaking. After this period, the solvent, along with the material fraction dissolved in it, was carefully removed, and the vials were desiccated in an oven at 80 °C for 20 h. They were then weighed to calculate the amount of lost components.
GPC analysis
The GPC analyses were performed on a Waters 600 E liquid chromatography system (Milford, MA, USA) connected to a HP1040 UV detector (Santa Clara, California, USA) set at 240 nm. The injection port was a Rheodyne loop valve (Rohnert Park, CA, USA) equipped with a 20-μL loop. The GPC-column system was composed of a sequence of 5 μm of 500 Ǻ Agilent PL gel (Santa Clara, CA, USA) and 5 μm of 104 Ǻ Agilent PL gel. The samples were dissolved in THF (1 mg/mL) and analyzed at a flow rate of 1 mL/min using THF as the eluent (Fluka 99.8%). The PL Polymer Standards of Polystyrene from Polymer Laboratories (Church Stretton, UK) were used for calibration.
DSC analysis
The DSC analysis was performed with a Mettler Toledo Stare thermal analysis system (Columbus, OH, USA) equipped with a liquid N2 low-temperature apparatus. The experiments were run under nitrogen atmosphere. Samples of selected polymers were sealed into 40-µL aluminum pans under dry conditions, cooled to -80 °C for 3 min, and then heated to 150 °C at 20 °C/min. The samples were kept at 150 °C for 3 min, cooled down to -80 °C at 20 °C/min, and then re-heated to 150 °C at the same rate. Indium was used as the standard for temperature and heat flow calibration. The Tg values were obtained at the mid-point of the step transition in the calorimetric curve. The complete thermograms are reported in the Appendix (Figs. S5 to S12).
RESULTS AND DISCUSSION
Improvements to the Lignin Epoxidation
Lignin epoxidation was performed according to a modified procedure, which was already published by the authors (Salanti et al. 2016a) and based on a simplification of an epoxidation procedure reported by Chen et al. (2016). This new strategy was preferred because it allows for the separation of high molecular weight fragments, and thus solely collected the lignin fraction suitable for further wet processing.
Table 1 summarizes the quantification of the epoxide groups, expressed as mmol/g, that was calculated from the quantitative 31P and 13C NMR analyses. For comparison, the same values obtained in a preceding publication were also reported (Salanti et al. 2016a). Epoxide groups quantified through 31P NMR spectroscopy (Fig. 1, middle) was designated as hydroxypropylic -OH to highlight the peculiar chemical structure that resulted from the reaction between the oxirane ring and phosphitylating reagent used for derivatization. After the glycidylation reaction, only a trace amount of free phenols was detected and a negligible difference in the 31P NMR quantification of the epoxides was observed between the old and new experimental procedures (2.25 mmol/g vs. 2.34 mmol/g, respectively). However, it is worth noting the impressive increase registered in the yield of LignEP with the new method that was employed (92% vs. 62% from the former method), which could have been ascribed to the formation of a minimal amount of water-soluble and cross-linked side-products. It is known that, depending upon the reaction conditions, epichlorohydrin can promote cross-linking reactions (Jyothi et al. 2006). The FT-IR analysis of the solid residue left after the acetone dissolution (spectra not reported) showed a different distribution in the intensities of the -OH groups stretching (approximately 3400 cm-1) and the C-O stretching of the epoxide rings (approximately 905 cm-1, 830 cm-1, and 750 cm-1) and aliphatic alcohols (approximately 1020 cm-1) when compared with the same peaks in the acetone-soluble LignEP spectrum. In particular, the presence of a minor amount of epoxide rings, along with a major relative abundance of hydroxyls, supported the hypothesis that crosslinking reactions occurred. Moreover, the observed decrease in acidic functionalities after the glycidylation reaction (Fig. 1, middle) was interpreted from the formation of condensation side-products formed by the reactions between the carboxylic and epoxide groups.
Table 1. Epoxide Group Quantification According to the 31P and 13C NMR Analyses
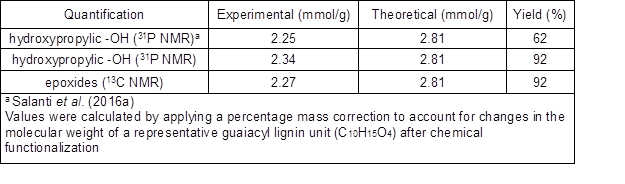
The amount of epoxide groups inserted on the lignin was also calculated by quantitative 13C NMR analysis (Supporting Information, Fig. S2) using epibromohydrin as the internal standard. The value obtained from this analysis agreed with the 31P NMR analysis (2.27 vs. 2.34, respectively). This double approach towards the quantification of the epoxides is particularly useful for rejecting the occurrence of hydrolysis phenomena under the new reaction conditions. In fact, 31P NMR analysis is unable to provide this kind of information because it relies on an indirect estimation based on a ring opening reaction. The quantification of epoxide groups through 31P NMR analysis, according to an already established procedure (Salanti et al. 2016a), is achieved by subtracting to the total amount of aliphatic hydroxyls detected after epoxidation from the amount of aliphatic hydroxyls in the unfunctionalized reference lignin (after a convenient mass correction). Thus, hydrolysis side-reactions produced a systematic overestimation of the epoxy moieties detected by the 31P NMR analysis, which was not found in the examined case.
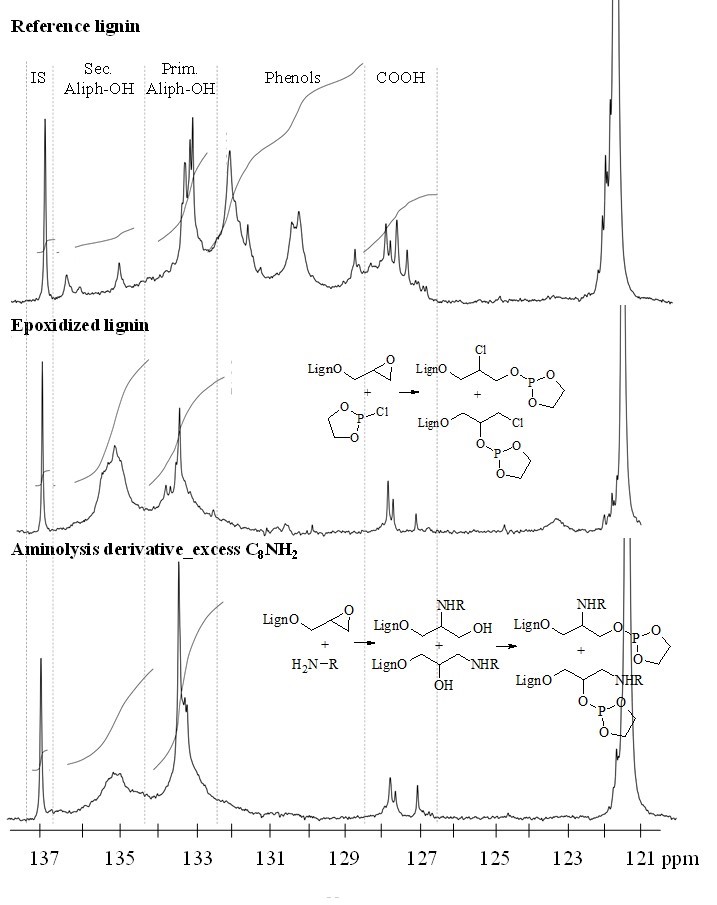
Fig. 1. 31P NMR spectra of the reference lignin, LignEP, and aminolysis lignin derivative that show the characteristic distribution of the primary and secondary aliphatic alcohols after the epoxidation and aminolysis reactions
Model Aminolysis Reaction
The progress of the aminolysis reaction on the LignEP was studied both under excess and stoichiometric amine conditions to assess the efficiency of the process. At first, the reaction in the presence of excess C8NH2 was investigated by FT-IR and quantitative 31P NMR analyses, and evaluated the amount of aliphatic -OH formed as a consequence of the ring-opening reaction. Figure 1 reports the 31P NMR spectra of the reference lignin, LignEP, and aminolysis lignin derivative after the reaction with 2-chloro-1,3,2-dioxaphospholane, which was used as a derivatizing agent to better resolve the primary and secondary aliphatic hydroxyls. After aminolysis, the amount of -OH moieties provided by the β-aminoalcohol were the same as the amount of aliphatic hydroxyls that naturally occur in pristine lignin. As a consequence of the increased molecular weight, a convenient mass correction should be applied to the amount of -OH groups that naturally occur in lignin before to compute the contribution of hydroxyls derived from the β-aminoalcoholic structures. The result, estimated at 2.21 mmol/g, showed that the experimentally calculated total amount of β-aminoalcohol was in good agreement with the theoretical value (1.99 mmol/g). The absence of residual glycidyl groups was verified through the FT-IR and 13C NMR analyses (Figs. S1 and S3, respectively).
The 31P NMR spectroscopy was also used to evaluate the occurrence of a preferred regioselectivity in the ring-opening reaction. In fact, the nucleophilic attack of the amino group on the oxirane rings can follow two different paths, which resulted in a β-aminoalcoholic structure bearing either a primary or secondary hydroxyl (see structures at the bottom of Fig. 1). It was determined that the ring-opening mechanism was not subject to a preferred regioselectivity because the secondary to primary aliphatic hydroxyls ratio was 0.92. However, it is possible that this trend could be intimately connected to the reaction conditions employed (amine concentration, in-dry environment).
Because it is a well-recognized fact that in traditional epoxy compositions each N-H moiety of an amino group reacts with an epoxy ring during the polyaddition process, the formation of the aminolysis derivative under stoichiometric conditions was also studied. The outcome of this reaction has never been investigated on lignin, and such chemical behavior is not at all predictable for a stiff, multifunctional, and tridimensional macromolecule like lignin. The LignEP was therefore reacted with 1-octylamine according to a 2:1 epoxy to amino group stoichiometric ratio, and the resulting polymer was analyzed. For comparison, the dendrimeric polymer obtained by reaction with excess C8NH2 that was previously discussed was considered. In both cases, the FT-IR analysis revealed the absence of oxirane rings (Fig. S1), and characteristic peaks of epoxide groups at approximately 905 cm-1, 830 cm-1, and 750 cm-1. Also, more intense absorptions related to C-H stretching of the amine chain were observed.
The 31P NMR analysis could not be performed on the stoichiometric derivative because of the substantial insolubility of the sample in the analytical solvent mixture. It was therefore decided to acetylate the derivative for characterization by GPC and 13C NMR analyses. Interestingly, the stoichiometric specimen was only completely solubilized in the acetylation mixture (pyridine-acetic anhydride = 1:1) after several hours (the specimen prepared in C8NH2 excess conditions readily dissolved). After recovery, it was found to be almost insoluble in THF (whereas the specimen prepared in C8NH2 excess conditions was thoroughly soluble) and only partially soluble in polar aprotic solvents that usually provide an efficient dissolution of lignin derivatives. In light of these qualitative observations, it was therefore inferred that each one of the N-H moieties of an amine group reacts with an epoxy unit on the lignin, and this stoichiometry was kept fixed for all of the resin preparations discussed in the following section.
Epoxy Resins Preparation and Characterization
Thermoset resins containing LignEP as a co-macromonomer combined with PEGEP and two different amines were prepared in accordance with the compositions reported in Table 2. The LignEP served both as a cross-linker and hard segment; the PEGEP acted as a soft segment to modulate the polymer characteristics; the DAD was used both as a tentative chain extender and co-operating cross-linker; and 1-octylamine was used as a pure chain extender. A number of PEGEP/LignEP equivalent ratios were investigated to cover a wide range of resin structures. This approach also attempted to tune the solubility properties of the resulting resin for analysis in solution. It was expected that a higher percentage of PEGEP in the composition would open up the polymer structure and decrease the cross-linking density, while increasing the solvent accessible surface area.
Table 2. Composition of the Reaction Mixtures Used to Prepare Lignin Cross-linked Epoxy Resins, Appearance after Curing, Glass Transition Temperature, and Percent of Soluble Material after Swelling in THF
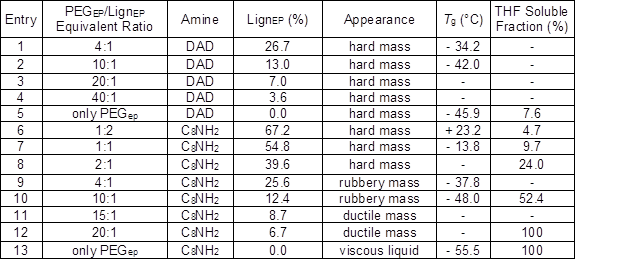
The epoxy/NH equivalent ratio was fixed at 1:1; Tg was measured for the second heating cycle for selected polymers analyzed by DSC; Hyphens indicate analyses were not done
At first, the LignEP was employed as an assistant cross-linker along with the di-functional amine DAD (Table 2; entries 1 to 4), which also served as a chain extender. However, the resulting resins were always recovered in the form of a hard mass that was completely insoluble in the analytical solvents, despite the large percent of PEGEP introduced into the formulation. This was also true for the blank sample that did not contain lignin in the composition (entry 5). Clearly, the role played by the DAD as a reticulating agent was too effective to neatly evaluate the performance of the lignin in the very same role. In spite of these results, the DSC analyses (thermograms provided in Figs. S7, S8, and S9) performed on the blank reference sample and two resins containing lignin (Table 2; entries 1, 2, and 5) showed that the thermal properties of the samples were affected by its presence. Higher Tg values were detected at larger lignin concentrations, which denoted a higher cross-linking degree that was apparently because of the presence of more than two reticulation points in the average LignEP macromolecule.
Therefore, DAD was substituted with C8NH2 to assess the ability of the lignin to act as a proper and unique cross-linking agent in the resin composition (Fig. 2). In this case, the amine, which is essential for the polyaddition reaction to take place, served only as a chain extender in its monofunctional form. It was observed that the physical appearance of the resin varied as a function of the lignin content (FT-IR spectra available in Fig. S4). For the lower PEGEP/LignEP ratios employed (Table 2; entries 6, 7, and 8), the result was a hard resin that was completely insoluble in any of the analytical solvents; for the medium PEGEP/LignEP ratios (Table 2; entries 9 and 10), a rubbery mass not impaired by the solvent treatment was recovered; and for the higher PEGEP/LignEP ratios (Table 2; entries 11 and 12), a ductile gel was obtained. This trend was clearly connected to the different degrees of reticulation of the various resin constituents. As further confirmation, the polymer recovered from the reference sample, composed only of PEGEP and C8NH2 (Table 2; entry 13), was a viscous liquid, as was expected because of the absence of any cross-linking reactions.
First, the cross-linking degree was assessed through a solvation test, where the percent of the material lost as a consequence of the solvent treatment was measured. Non-crosslinked material diffuses into the excess solvent, which potentially leaves a reticulated tridimensional structure swollen by the solvent. A proper swelling test, including the evaluation of the volumetric changes to the resin, could not be undertaken because of issues related to the preparation of regularly-shaped specimens. According to the values reported in Table 2 (last column), an evident trend was observed that connected the amount of dissolved material to the expected degree of reticulation. In fact, the whole specimen was instantly dissolved in THF for the blank (entry 13) and high PEGEP/LignEP ratio samples (entries 11 and 12). After increasing the PEGEP/LignEP ratio, the amount of solubilized polymer decreased accordingly, and reached remarkable values of 9.7% and 4.7% for the 1:1 and 1:2 PEGEP/LignEP stoichiometric ratios, respectively. It should be noted that these values were virtually the same as the ones obtained for the fully cross-linked blank sample involving the di-functional amine DAD in the composition (entry 5), which confirmed the formation of a highly interconnected structure.
The DSC analysis of select samples (entries 6, 7, 9, 10, and 13) corroborated that there was a reticulating action exerted by the lignin among the resin components. The majority of the thermograms (some provided as supplementary material in Figs. S8, S9, S10, S11, and S12) exhibited an evaporation phenomenon during the first heating cycle around 80 °C, which was not observed during the second heating cycle, which suggested the presence of residual solvent (THF) in the samples. More interestingly, the Tg of the examined polymers was appreciably influenced by the presence of LignEP. According to Table 2, a lower Tg was detected for the reference non-reticulated resin (-55.5 °C, entry 13), whereas the formulations including lignin achieved a Tg value that increased proportional to the lignin content. Accordingly, a Tg value equal to -48.0 °C was detected for the 10:1 PEGEP/LignEP equivalent ratio (entry 10); a Tg of -37.8 °C was observed for the 4:1 PEGEP/LignEP composition (entry 9); and a Tg value of -13.8 °C was computed for the 1:1 PEGEP/LignEP equivalent ratio (entry 7). In contrast with the above listed specimens, whose Tg values were all negative, the thermogram of the polymer obtained with a 1:2 PEGEP/LignEP equivalent ratio revealed a positive Tg (23.2 °C) as a consequence of the increasing contribution of lignin to the overall thermal properties of the resin (average Tg values of alkaline lignins are around 150 °C). This trend was a clear consequence of the cross-linking action exerted by the LignEP macromolecule and proof of the reduced mobility of the polymeric chains. Moreover, the exothermic crystallization (observed around -30 °C) became less defined during the cooling cycle with an increasing lignin content. This result indicates the molecular dispersion of the components.
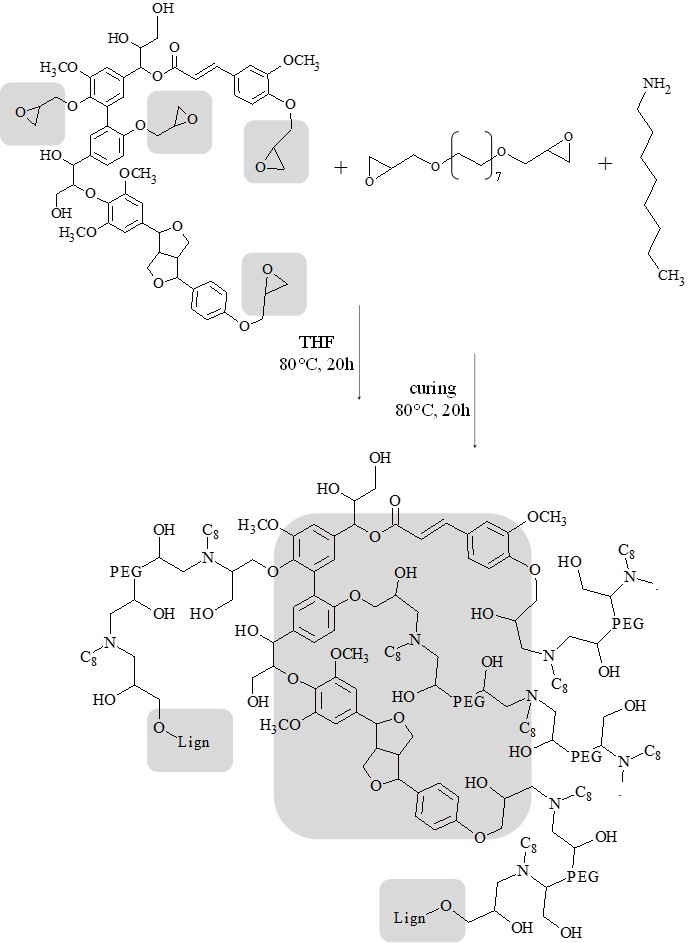
Fig. 2. Reactants, products, and experimental conditions used for the preparation of lignin cross-linked epoxy resins (representative fragment of LignEP, Mn ≈ 3000, epoxide content 2.27 mmol/g; PEGEP, Mn ≈ 500, functionality 2; 1-octylamine, MW = 129.2 g/mol, functionality 2). Reticulation points are highlighted in grey
To evaluate the development of the reticulating process, the effect of time on the molecular weight distribution of the entry 10 resin was analyzed. The GPC monitoring was performed over the first 8 h of reaction; after this period, the resulting resin was insoluble in THF for the most part, which indicated a further increase in the average molecular weight. The GPC chromatograms of the most indicative samples, along with reference chromatograms of the LignEP and blank sample (Table 2; entry 13), are displayed in Fig. 3. Over the examined reaction period, the average molecular weight progressively increased, and gradually consumed the free LignEP fraction. It should be noted that the peak molecular weight shifted from approximately 1000 g/mol at 0 h (reference LignEP) to 15000 g/mol after 8 h. Moreover, the molecular weight distribution detected for the last sample evidently shifted towards a higher molecular weight than the corresponding reference sample, which further confirmed the cross-linking action of the LignEP. Simultaneously, because of the adopted strategy involving the use of a monofunctional amine, it was further verified that both the N-H of the amine group reacted during the polyaddition process.
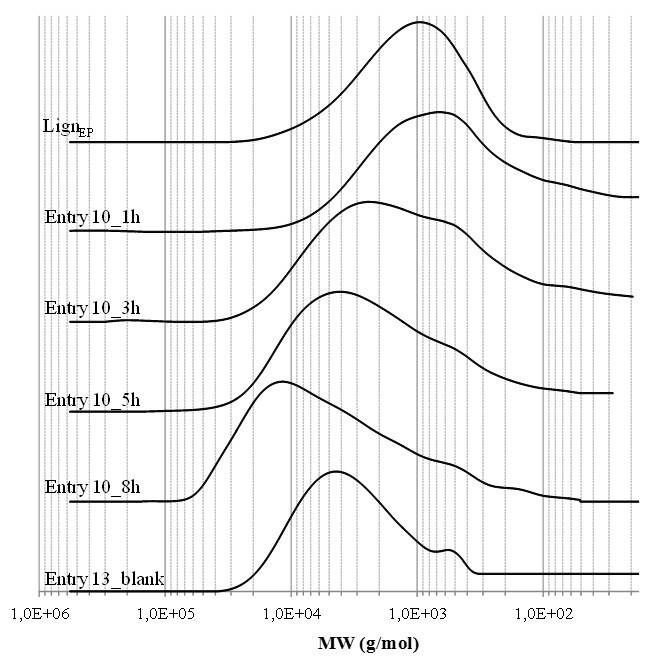
Fig. 3. Molecular weight profile evolution after 1 h (Entry 10_1h), 3 h (Entry 10_3h), 5 h (Entry 10_5h), and 8 h (Entry 10_8h) of reaction. At the end of the established reaction period (20 h), the mixture was a THF-insoluble gel. The molecular weight distributions of the LignEP and corresponding benzoylated blank sample (Entry 13_blank) were reported as references.
CONCLUSIONS
- Epoxidized lignin was successfully employed as the unique reticulating agent in the preparation of epoxy resins.
- Epoxidized lignin could partially or totally substitute petroleum-derived cross-linkers and/or rigid segments in the synthesis of epoxy resins.
- Because of its stiff aromatic structure, the LignEP was able to serve as a rigid segment, and comprehensively improved the thermal properties of the resulting polymer, which was confirmed by the DSC analysis.
- Different PEGEP/LignEP equivalent ratios were investigated to tailor the resin characteristics.
- The properties of the obtained polymers, which contained up to 67% LignEP, demonstrated the possibility of building a networked structure relying only on the reticulating capability of lignin. In fact, the resin composition exploited a monofunctional amine that acted only as a chain extender and not as a cross-linking agent because of its structure.
REFERENCES CITED
Aouf, C., Lecomte, J., Villeneuve, P., Dubreucq, E., and Fulcrand, H. (2012). “Chemo-enzymatic functionalization of gallic and vanillic acids: Synthesis of bio-based epoxy resins prepolymers,” Green Chem. 14(8), 2328-2336. DOI: 10.1039/C2GC35558B
Aouf, C., Le Guernevé, C., Caillol, S., and Fulcrand, H. (2013). “Study of the o-glycidylation of natural phenolic compounds. The relationship between the phenolic structure and the reaction mechanism,” Tetrahedron 69(4), 1345-1353. DOI: 10.1016/j.tet.2012.079
Asada, C., Basnet, S., Otsuka, M., Sasaki, C., and Nakamura, Y. (2015). “Epoxy resin synthesis using low molecular weight lignin separated from various lignocellulosic materials,” Int. J. Biol. Macromol. 74, 413-419. DOI: 10.1016/j.ijbiomac.2014.12.039
Auvergne, R., Caillol, S., David, G., Boutevin, B., and Pascault, J.-P. (2014). “Biobased thermosetting epoxy: Present and future,” Chem. Rev. 114(2), 1082-1115. DOI: 10.1021/cr3001274
Baroncini, E. A., Yadav, S. K., Palmese, G. R., and Stanzione, J. F. (2016). “Recent advances in bio-based epoxy resins and bio-based epoxy curing agents,” J. Appl. Polym. Sci. 133(45). DOI: 10.1002/app.44103
Becker, K., Güen, T., Seiwert, M., Conrad, A., Pick-Fuß, H., Müller, J., Wittassek, M., Schulz, C., and Kolossa-Gehring, M. (2009). “GerES IV: Phthalate metabolites and bisphenol A in urine of German children,” Int. J. of Hyg. Envir. Heal. 212(6), 685-692. DOI: 10.1016/j.ijheh.2009.08.002
Castan, P. (1943). “Process of preparing synthetic resins,” U.S. Patent No. 2324483.
Chen, C., Zhu, M., Li, M., Fan, Y., and Sun, R.-C. (2016). “Epoxidation and etherification of alkaline lignin to prepare water-soluble derivatives and its performance in improvement of enzymatic hydrolysis efficiency,” Biotechnol. Biofuels 9, 87-101. DOI: 10.1186/s13068-016-0499-9
Dodds, E. C., and Lawson, W. (1936). “Synthetic estrogenic agents without the phenanthrene nucleus,” Nature 137(3476), 996. DOI: 10.1038/137996a0
Ferdosian, F., Yuan, Z., Anderson, M., and Xu, C. C. (2014). “Synthesis of lignin-based epoxy resins: Optimization of reaction parameters using response surface methodology,” RSC Adv. 4(60), 31745-31753. DOI: 10.1039/C4RA03978E
Ferdosian, F., Yuan, Z., Anderson, M., and Xu, C. C. (2015). “Sustainable lignin-based epoxy resins cured with aromatic and aliphatic amine curing agents: Curing kinetics and thermal properties,” Thermochim. Acta 618, 48-55. DOI: 10.1016/j.tca.2015.09.012
Ferdosian, F., Yuan, Z., Anderson, M., and Xu, C. C. (2016). “Synthesis and characterization of hydrolysis lignin-based epoxy resins,” Ind. Crop. Prod. 91, 295-301. DOI: 10.1016/j.indcrop.2016.07.020
Fourcade, D., Ritter, B. S., Walter, P., Schönfeld, R., and Mülhaupt, R. (2013). “Renewable resource-based epoxy resins derived from multifunctional poly(4-hydroxybenzoates),” Green Chem. 15(4), 910-918. DOI: 10.1039/C3GC37088G
Gandini, A. (2010). “Epoxy polymers based on renewable resources” in: Epoxy Polymers: New Materials and Innovations, J.-P. Pascault and R. J. J. Williams (eds.), Wiley-VCH, Weinheim, Germany.
Hernandez, E. D., Bassett, A. W., Sadler, J. M., La Scala, J. J., and Stanzione, J. F. (2016). “Synthesis and characterization of bio-based epoxy resins derived from vanillyl alcohol,” ACS Sustain. Chem. Eng. 4(8), 4328-4339. DOI: 10.1021/acssuschemeng.6b00835
Hofmann, K., and Glasser, W. G. (1993). “Engineering plastics from lignin. 21. Synthesis and properties of epoxidized lignin-poly (propylene oxide) copolymers,” J. Wood Chem. Technol. 13(1), 73-95. DOI: 10.1080/02773819308020508
Jordan, J. M., Michelotti, F. W., Pearce, E. M., and Zief, M. (1970). “Novel thermosetting epoxy resins based on pentaerythritol,” in: Epoxy Resins, H. Lee (ed.), American Chemical Society, Washington, DC, pp. 1-7.
Jyothi, A. N., Moorthy, S. N., and Rajasekharan, K. N. (2006). “Effect of cross-linking with epichlorohydrin on the properties of cassava (Manihot esculenta Crantz) starch,” Starch-Stärke 58(6), 292-299. DOI: 10.1002/star.200500468
Kaiho, A., Mazzarella, D., Satake, M., Kogo, M., Sakai, R., and Watanabe, T. (2016). “Construction of the di(trimethylolpropane) cross linkage and the phenylnaphthalene structure coupled with selective β-O-4 bond cleavage for synthesizing lignin-based epoxy resins with a controlled glass transition temperature,” Green Chem. 18(24), 6526-6535. DOI: 10.1039/C6GC02211A
Koike, T. (2012). “Progress in development of epoxy resin systems based on wood biomass in Japan,” Polym. Eng. Sci. 52(4), 701-717. DOI: 10.1002/pen.23119
Ng, F., Couture, G., Philippe, C., Boutevin, B., and Caillol, S. (2017). “Bio-based aromatic epoxy monomers for thermoset materials,” Molecules 22(1), 149-196. DOI: 10.3390/molecules22010149
Park, S.-J., Jin, F.-L., and Lee, J.-R. (2004). “Synthesis and thermal properties of epoxidized vegetable oil,” Macromol. Rapid Comm. 25(6), 724-727. DOI: 10.1002/marc.200300191
Salanti, A., Zoia, L., and Orlandi, M. (2016a). “Chemical modifications of lignin for the preparation of macromers containing cyclic carbonates,” Green Chem. 18(14), 4063-4072. DOI: 10.1039/C6GC01028H
Salanti, A., Zoia, L., Zanini, S., and Orlandi, M. (2016b). “Synthesis and characterization of lignin–silicone hybrid polymers as possible consolidants for decayed wood,” Wood Sci. Technol. 50(1), 117-134. DOI: 10.1007/s00226-015-0772-2
Salanti, A., Zoia, L., Mauri, M., and Orlandi, M. (2017). “Utilization of cyclocarbonated lignin as bio-based cross-linker for the preparation of poly(hydroxy urethane)s,” RSC Adv. 7(40), 25054-25065. DOI: 10.1039/C7RA03416D
Santacesaria, E., Tesser, R., Di Serio, M., Casale, L., and Verde, D. (2009). “New process for producing epichlorohydrin via glycerol chlorination,” Ind. Eng. Chem. Res. 49(3), 964-970. DOI: 10.1021/ie900650x
Sasaki, C., Wanaka, M., Takagi, H., Tamura, S., Asada, C., and Nakamura, Y. (2013). “Evaluation of epoxy resins synthesized from steam-exploded bamboo lignin,” Ind. Crop. Prod. 43, 757-761. DOI: 10.1016/j.indcrop.2012.08.018
Shibata, M., and Nakai, K. (2010). “Preparation and properties of biocomposites composed of bio-based epoxy resin, tannic acid, and microfibrillated cellulose,” J. Polym. Sci. Poly. Phys. 48(4), 425-433. DOI: 10.1002/polb.21903
Shibata, M., Yoshihara, S., Yashiro, M., and Ohno, Y. (2013). “Thermal and mechanical properties of sorbitol-based epoxy resin cured with quercetin and the biocomposites with wood flour,” J. Appl. Polym. Sci. 128(5), 2753-2758. DOI: 10.1002/app.38438
Simionescu, C., Rusan, V., Turta, K., Bobcova, S., Macoveanu, M., Cazacu, G., and Stoleriu, A. (1993). “Synthesis and characterization of some iron-lignosulfonate-based lignin-epoxy resins,” Cell. Chem. Technol. 27(6), 627-644.
Spectral Database of Organic Compounds SDBS (2017). “SDBS compounds and spectral search,” National Institute of Advanced Industrial Science and Technology, (http://sdbs.db.aist.go.jp), Accessed 14 June 2017.
Supanchaiyamat, N., Shuttleworth, P. S., Hunt, A. J., Clark, J. H., and Matharu, A. S. (2012). “Thermosetting resin based on epoxidised linseed oil and bio-derived crosslinker,” Green Chem. 14(6), 1759-1765. DOI: 10.1039/C2GC35154D
Upton, B. M., and Kasko, A. M. (2016). “Strategies for the conversion of lignin to high-value polymeric materials: Review and perspective,” Chem. Rev. 116(4), 2275-2306. DOI: 10.1021/acs.chemrev.5b00345
Wan, J., Gan, B., Li, C., Molina-Aldareguia, J., Li, Z., Wang, X., and Wang, D.-Y. (2015). “A novel biobased epoxy resin with high mechanical stiffness and low flammability: Synthesis, characterization and properties,” J. Mater. Chem. A 3(43), 21907-21921. DOI: 10.1039/C5TA02939B
Xin, J., Li, M., Li, R., Wolcott, M. P., and Zhang, J. (2016). “Green epoxy resin system based on lignin and tung oil and its application in epoxy asphalt,” ACS Sustain Chem. Eng. 4(5), 2754-2761. DOI: 10.1021/acssuschemeng.6b00256
Zhao, B., Chen, G., Liu, Y., Hu, K., and Wu, R. (2001). “Synthesis of lignin base epoxy resin and its characterization,” J. Mater. Sci. Lett. 20(9), 859-862. DOI: 10.1023/A:1010975132530
Article submitted: October 17, 2017; Peer review completed: January 13, 2018; Revised version received: January 30, 2018; Accepted: February 1, 2018; Published: February 8, 2018.
DOI: 10.15376/biores.13.2.2374-2396
APPENDIX
Supplementary Data
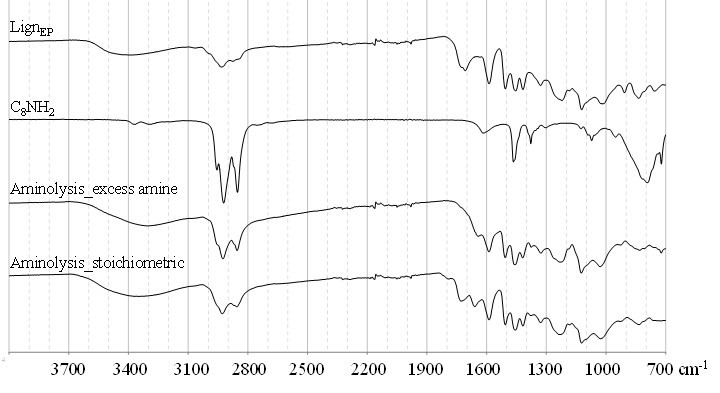
Fig. S1. ATR-FTIR analysis of aminolysis derivatives
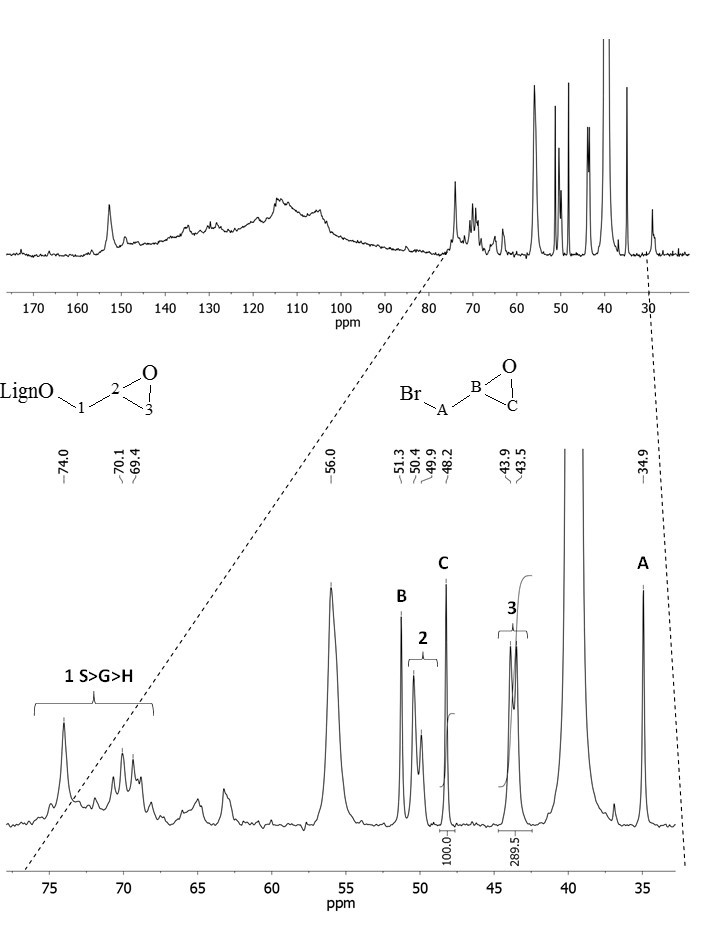
Fig. S2. Quantitative 13C NMR spectra of epoxidized lignin
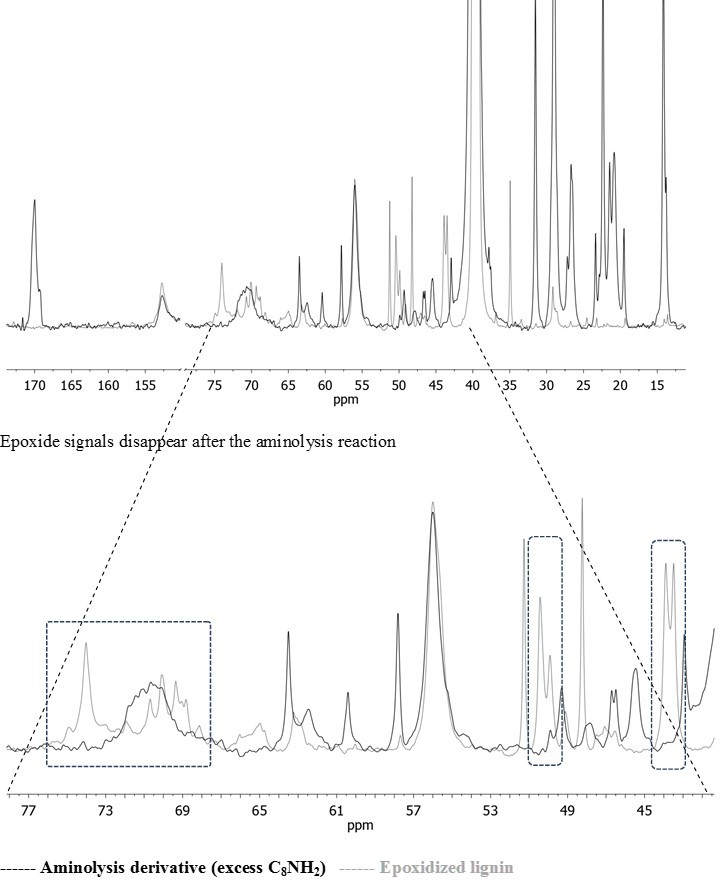
Fig. S3. Overlapped 13C NMR Spectra of epoxidized and aminolysis lignin
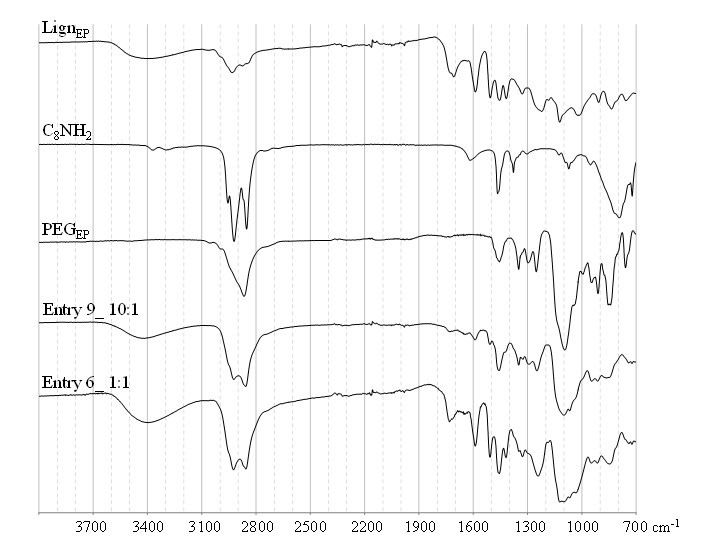
Fig. S4. ATR-FTIR analysis of representative resins specimens
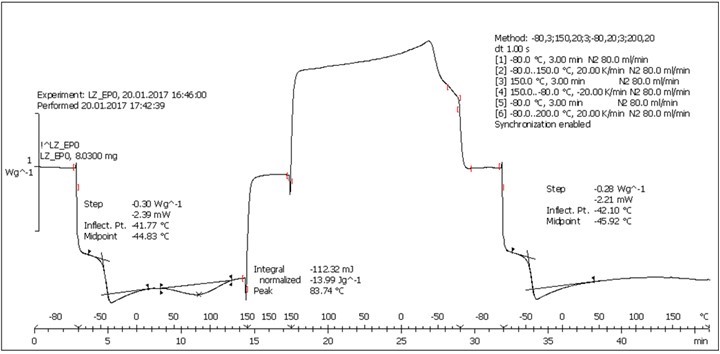
Fig. S5. DSC analysis: Table 2, Entry 5 – Blank reference (PEGEP + DAD)
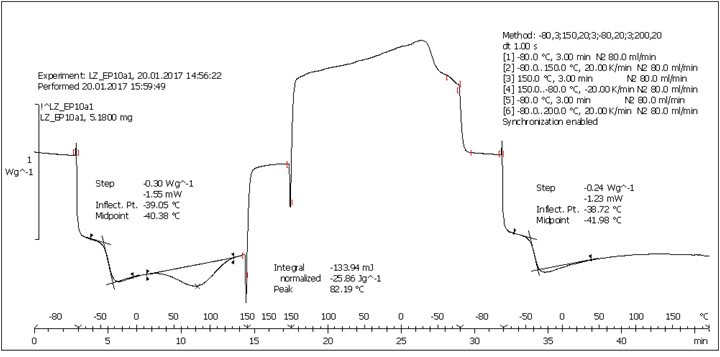
Fig. S6. DSC analysis: Table 2, Entry 2 – PEGEP/LIGNEP 10:1 (LIGNEP + PEGEP + DAD)
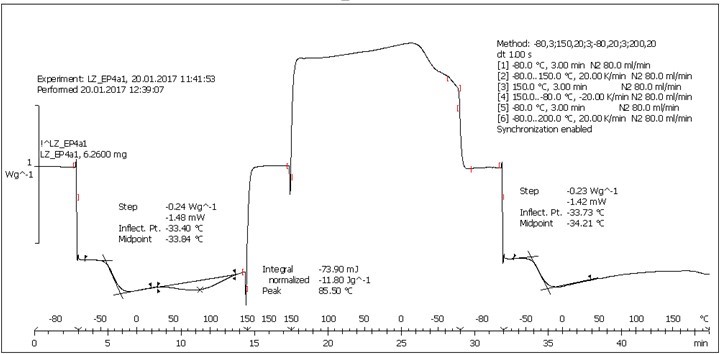
Fig. S7. DSC analysis: Table 2, Entry 1 – PEGEP/LIGNEP 4:1 (LIGNEP + PEGEP + DAD)
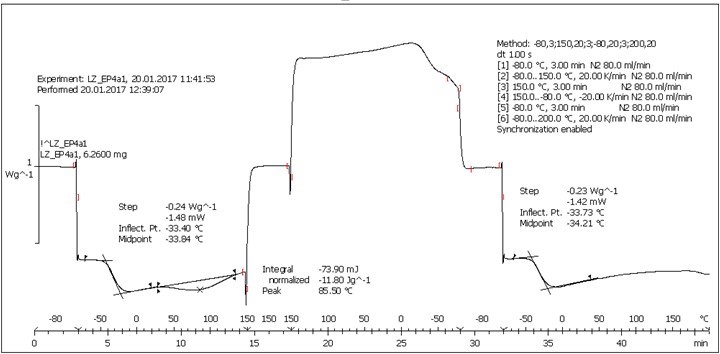
Fig. S8. DSC analysis: Table 2, Entry 12 – Blank reference (PEGEP + C8NH2
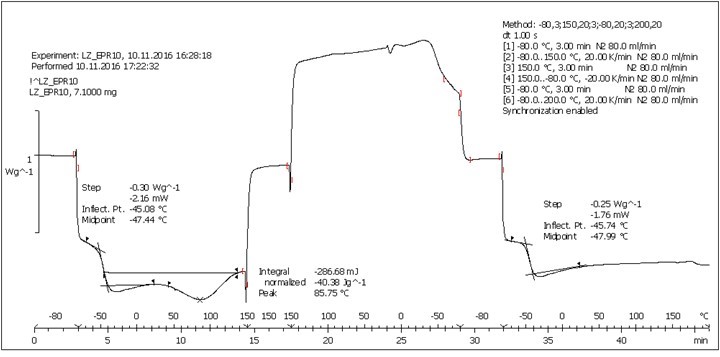
Fig. S9. DSC analysis: Table 2, Entry 10 – PEGEP/LIGNEP 10:1 (LIGNEP + PEGEP + C8NH2)
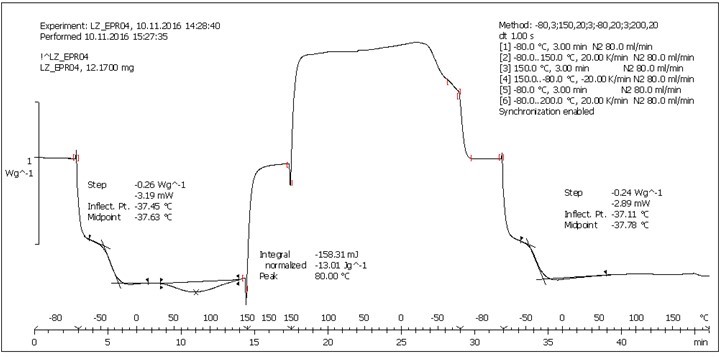
Fig. S10. DSC analysis: Table 2, Entry 9 – PEGEP/LIGNEP 4:1 (LIGNEP + PEGEP + C8NH2)
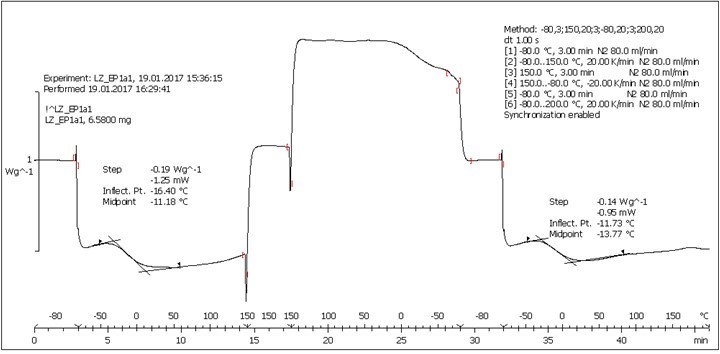
Fig. S11. DSC analysis: Table 2, Entry 7 – PEGEP/LIGNEP 1:1 (LIGNEP + PEGEP + C8NH2)
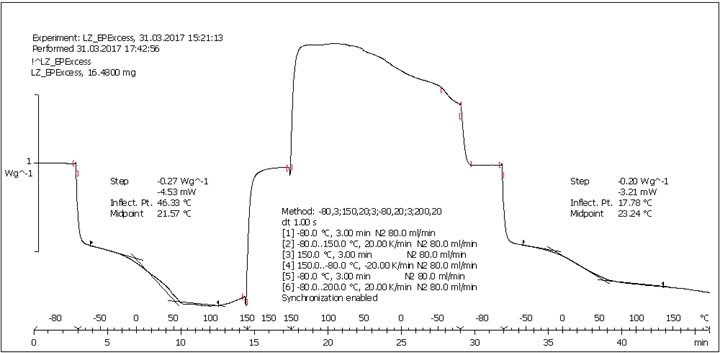
Fig. S12. DSC analysis: Table 2, Entry 6 – PEGEP/LIGNEP 1:2 (LIGNEP + PEGEP + C8NH2)