
Abstract
Lignin is one of the most important and widespread carbon sources on Earth. Significant amounts of lignin are delivered to the market by pulp mills and biorefineries, and there have been many efforts to develop routes for its valorization. Over the years, lignin has been used to produce biobased chemicals, materials, and advanced biofuels on the basis of its variable functionalities and physicochemical properties. Today, lignin’s applications are still limited by its heterogeneity, variability, and low reactivity. Thus, modification technologies have been developed to allow lignin to be suitable for a wider range of attractive industrial applications. The most common modifications used for this purpose include amination, methylation, demethylation, phenolation, sulfomethylation, oxyalkylation, acylation or esterification, epoxidation, phosphorylation, nitration, and sulfonation. This article reviews the chemistry involved in these lignin modification technologies, discussing their effect on the finished product while presenting some market perspectives and future outlook to utilize lignin in sustainable biorefineries.
Download PDF
Full Article
Lignin Functionalization Strategies and the Potential Applications of Its Derivatives – A Review
Maria Juliane Suota,a Débora Merediane Kochepka,a Marlon Gualberto Ganter Moura,a Cleverton Luiz Pirich,a Mailson Matos,b Washington Luiz Esteves Magalhães,b,c and Luiz Pereira Ramos a,*
Lignin is one of the most important and widespread carbon sources on Earth. Significant amounts of lignin are delivered to the market by pulp mills and biorefineries, and there have been many efforts to develop routes for its valorization. Over the years, lignin has been used to produce biobased chemicals, materials, and advanced biofuels on the basis of its variable functionalities and physicochemical properties. Today, lignin’s applications are still limited by its heterogeneity, variability, and low reactivity. Thus, modification technologies have been developed to allow lignin to be suitable for a wider range of attractive industrial applications. The most common modifications used for this purpose include amination, methylation, demethylation, phenolation, sulfomethylation, oxyalkylation, acylation or esterification, epoxidation, phosphorylation, nitration, and sulfonation. This article reviews the chemistry involved in these lignin modification technologies, discussing their effect on the finished product while presenting some market perspectives and future outlook to utilize lignin in sustainable biorefineries.
Keywords: Lignin functionalization; Derivatized lignin; Biobased products; Value-added materials
Contact information: a: Department of Chemistry, Federal University of Paraná (UFPR), 81531-980 Curitiba, PR, Brazil; b: Graduate Program in Materials Science and Engineering; Federal University of Paraná (UFPR), 81531-980 Curitiba, PR, Brazil; c: Embrapa Florestas, 83411-000 Colombo, PR, Brazil;
* Corresponding author: luiz.ramos@ufpr.br
GRAPHICAL ABSTRACT
INTRODUCTION
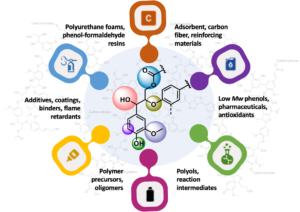
Since pulping and paper industries were established at the end of the nineteenth century, a massive amount of residual lignin has been incinerated to recover chemicals and produce power and steam at the pulp mill sites. Such processing recovers only the fuel-equivalent value of the lignin. However, lignin may be a promising source of biobased materials such as polyols, antioxidants, binders, coatings, reinforcing agents, and biofuels. Besides being one of the most important and widespread carbon source on Earth, lignin is capable of delivering renewable aromatic compounds on an extensive scale (Bajwa et al. 2019). Pulp mills demonstrate an increasing interest in alternative lignin applications to diversify their product portfolio and generate additional sources of revenue. Other drivers to this end are improvements in pulping efficiency, the development of cost-effective technologies for lignin isolation, and the possibility of using underutilized biomass such as branches and bark for cogeneration. With this, in the mid-to-long term, more lignin will be made available for conversion to biobased fuels, chemicals and materials (Dessbesell et al. 2020).
Lignin is a natural macromolecule deposited in the plant cell wall as a result of condensation reactions involving phenylpropanoid radicals generated from p-coumaryl, coniferyl, and sinapyl alcohols during the lignification process (Figs. 1A, B). These cinnamic alcohols are the basis of the primary lignin structure, forming monolignols that are named p-hydroxyphenyl (H), guaiacyl (G), and syringyl (S) units, respectively. These are distributed in variable ratios in the plant kingdom (Fig. 1C).

Fig. 1. Phenylpropanoid structure (A), types of monolignol precursors and their appearance in the lignin structure (B), and lignin types that are found in the plant kingdom (C)
Gymnosperm (softwoods) lignins have more than 90% of G units and low levels of H units in their structure. By contrast, angiosperm dicot (hardwoods) lignins contain a mixture of S and G units in different ratios, while angiosperm monocot (grasses or herbaceous species) lignins contain the three types of lignin monomers in variable quantities (Boerjan et al. 2003). In general, softwood species have lignin contents slightly higher than those of hardwood species. For instance, Mutturi et al. (2014) reported acid-insoluble (Klason) lignin contents in the range of 16.7 to 24.9 wt% and 27.1 to 31.4 wt% for seven hardwood and eight softwood species, respectively.
Sulfite, soda, kraft, and organosolv pulping technologies (please refer to the next section for basic descriptions) were developed to remove lignin from lignocellulosic materials (Hu et al. 2018). The industrial interest in these technical lignins has increased considerably over the past years, prompting people at companies and research institutes to find cost-effective strategies to isolate lignin from black liquors, spent liquors, and process slurries. After its isolation, lignin can be applied as a macromolecule or source of monomers for organic synthesis, with the chemical functionality resulting from separation processes or further chemical modification (Schutyser et al. 2018). However, the inherent heterogeneity of lignin is potentially increased during isolation, and its high degree of structural variability is considered a bottleneck for its valorization. At first glance, the lack of regularity might hinder the control and final quality of the lignin derivatives. By contrast, the full knowledge of lignin structure allows for its selective modification toward unique properties that enlarge the portfolio of possible lignin applications (Eraghi Kazzaz et al. 2019).
The development of alternative lignin applications is limited by its heterogeneity, variability, and low reactivity (Bajwa et al. 2019). Thereby, lignin fractionation, assisted or not by modification strategies such as amination, demethylation, demethoxylation, phenolation, sulfonation, methylation, and depolymerization, among others, are needed to tailor lignin into specific structure and properties. This review dwells with (I) the chemical properties of currently available lignins, (II) the reactions that lignins are prone to undertake, (III) the main structures and properties resulting after modification, and (IV) the targeted applications for each type of modified lignin.
Lignin Production Processes and Types of Technical Lignins
Brauns (1939) defined native lignin as the lignin isolated in such way that the isolation process does not cause any change in its structure and chemical composition. Therefore, lignins that incorporate features from the isolation process cannot be named native. This applies to technical lignins derived from sulfite (lignosulfonate), soda, kraft, organosolv pulping, and those originated from acid hydrolysis, enzyme treatments and, extraction procedures using ionic liquids and deep eutectic solvents.
Sulfite pulping, the first established chemical route to produce wood pulp in large scale, employs sulfite and bisulfite salts to obtain cellulose from lignocellulosic materials. As a result of sulfite pulping, lignosulfonates are produced by sulfonation of the alpha carbon atom, making lignin water-soluble and higher in its average molecular mass (Li and Takkellapati 2018). Today, due to its low efficiency in chemical recovery and high environmental impact to produce pulps with relatively low fiber strength for paper making, sulfite pulping accounts for less than 10% of the pulp mills worldwide. However, the interest in lignosulfonates grew over time because lignosulfonates are an important bulk material for vanillin production, in addition to applications as a plasticizer for concrete, flocculating agent, metal adsorbent, binder for composites, and dust suppressant (Aro and Fatehi 2017).
Kraft pulping is the leading pulping process worldwide. Sodium hydroxide and sodium sulfide act on lignocellulosic materials to release cellulose fibers, and a severely depolymerized lignin is recovered in a liquid phase named black liquor (Demuner et al. 2019). This process is advantageous due to the chemical recovery step, whereby the lignin-containing black liquor is concentrated and burned while reagents of the spent liquor are recovered and reactivated. Just 2% of kraft lignin is designated for applications other than burning. However, the composition of this renewable aromatic carbon source and the potential of kraft pulping to deliver vast amounts of lignin to the market have incentivized the use of kraft lignin as a source of biobased chemicals, fuels, and advanced materials (Hu et al. 2018; Li and Takkellapati 2018). Kraft pulping results in changes to lignin’s structure and composition. Sulfur is introduced as thiol groups, and residual carbohydrates (mostly from hemicelluloses) remain as contaminants in different levels. Besides, aryl-ether bonds are broken, and condensation reactions occur primarily through C5 of the aromatic ring of guaiacyl substructures (Demuner et al. 2019).
Soda pulping is very similar to kraft pulping except for the absence of Na2S in the pulping liquor. Most soda pulp mills were converted to kraft pulping when the latter was made commercially available. Nevertheless, soda pulping is more suitable for non-woody biomasses due to their lower lignin content and higher accessibility to the impregnating chemicals. The resulting soda lignin is free from sulfur despite being as condensed as kraft lignin. In the soda pulping process, anthraquinone (AQ) can be used as an additive that protects carbohydrates against degradation and improves pulping efficiency (Francis et al. 2008). AQ is active in the reductive cleavage of ether bonds and produces soda-AQ lignins deficient in -O-4′ linkages. However, the industrial use of AQ has been banned in several countries due to its toxicity and harsh environmental impact (Hart and Rudie 2014).
Delignification with organic solvents (organosolv pulping) is another example of sulfur-free pulping technologies. It combines solvent systems (usually a mixture of alcohol and water), temperature, pressure, and sometimes the use of an exogenous acid catalyst for the selective removal of hemicelluloses and lignin. Organosolv lignins are recovered by precipitation from the spent liquor in high-quality, since they do not contain ash and sulfur. Also, these lignins usually have higher apparent molecular masses, lower dispersities, and lower condensation degrees compared to kraft and sulfite lignins. These characteristics make organosolv lignins interesting for conversion into chemicals and value-added products (Li and Takkellapati 2018). Also, organosolv pulping is less polluting than kraft and sulfite processes, and it does not release foul odors. Besides, the solvents used in the extraction procedure can be recyclable and renewable, such as in the case of ethanol and water.
Another emergent and large-scale lignin source is hydrolysis lignin (HL) (Bajwa et al. 2019). Cellulosic ethanol mills generate several tons of lignin as a coproduct from pretreatment and enzymatic hydrolysis. In the end, a solid residue containing 65 to 80% lignin is obtained, with its chemistry strongly dependent on the technique used for pretreatment and some nitrogen content due to enzyme adsorption (Li and Takkellapati 2018; Bajwa et al. 2019).
In addition to the aforementioned technical lignins, the ionosolv pulping technology delivers ionic liquid (IL) extracted lignins with properties and composition that depend on the applied process parameters (Rashid et al. 2021). Some ILs are very selective in lignin removal, while others remove plant polysaccharides or lead to complete lignocellulose dissolution. Ionosolv is similar to organosolv in operating temperatures, and the solvent applied is an IL with the ability to dissolve lignin (Schutyser et al. 2018). Lignin can be precipitated from this liquor, and its properties strongly depend on the severity used for delignification (Rashid et al. 2021).
Other green chemicals such as deep eutectic solvents and supercritical carbon dioxide (scCO2) have also been used for lignin isolation (Chen and Wan 2018; Jiang et al. 2020). These processes are friendly alternatives compared to isolation routes that employ high charges of harsh inorganic chemicals (Ramos et al. 2020). The scCO2 medium combined with a co-solvent such as ethanol and water penetrates the fiber cell walls, facilitating hemicellulose hydrolysis and lignin removal (Jiang et al. 2020). Carbonic acid formed from CO2 and water at high temperatures and pressures assists the partial depolymerization of hemicelluloses and lignin, along with other organic acids that are released in situ (e.g., acetic acid). Lignin isolated by scCO2 may be rich in ether linkages, making it suitable to produce polyols for further processing into polyurethane foams and adhesives (Jiang et al. 2020).
The increasing interest in lignin chemistry encouraged the development of lignin isolation strategies from pulping liquors, such as LignoForceTM and LignoBoostTM. Both technologies employ acid precipitation using carbon dioxide (CO2) for recovering lignin from the kraft pulping black liquor. Lignin is precipitated, washed, and dried to yield a solid powder with low carbohydrate and ash contents. The advantage of LignoForceTM over LignoBoostTM and conventional acid precipitation is a previous black liquor oxidization step that improves lignin filterability, reduces chemical consumption and minimizes the emission of volatile sulfur derivatives throughout the production process (Kouisni et al. 2016).
Excellent reviews can be found in the literature containing detailed information about pulping and pretreatment processes, lignin chemistry, and the prospects for use of technical lignins (Li and Takkellapati 2018; Schutyser et al. 2018; Bajwa et al. 2019).
LIGNIN FUNCTIONALIZATION
The presence of multiple functional groups represents the level of lignin’s complexity, besides its macromolecular structure. Functional groups such as alcohols, phenols, and ethers are distributed throughout the phenylpropanoid structure, decorating the biopolymer and making it susceptible to chemical modification (Fig. 2).

Fig. 2. Reactive sites of a lignin model structure and reactions that may be used to develop ideal properties for specific applications

Fig. 3. Main lignin transformations in aromatic and aliphatic (side-chain) moieties
Likewise, the original functional groups themselves may act as acids, nucleophiles, or electrophiles against a series of organic compounds. As a result, many transformations in lignin structure are possible, providing an increase or decrease in molecular mass distribution, an increase in reactivity due to the augmentation of highly reactive functional groups (e.g., phenolic and aliphatic hydroxyls), and an improvement of its compatibility with some synthetic polymeric matrices (Fig. 3). These include amination, methylation, demethylation, phenolation, sulfomethylation, oxyalkylation, acylation or esterification, epoxidation, phosphorylation, nitration, and sulfonation. Besides, improved solubility and thermal stability may be achieved, depending on the new functional groups attached to the lignin macromolecule.
Amination
Nitrogen is not an abundant element in lignin composition, as demonstrated by its elemental analysis (Wang et al. 2014; Ge et al. 2015). However, the use of amines to graft nitrogen into the lignin structure seems to be an exciting option for adding reactive sites capable of being used as intermediaries for lignin functionalization. In acidic conditions, amino groups are ionizable and positively charged, making lignin highly reactive in aqueous media (Wang et al. 2018a). Amino groups may also convert hydrophobic technical lignins such as kraft lignin into a highly hydrophilic material, improving its foamability, emulsifying properties, aging resistance, and mechanical strength (Liu et al. 2016). Also, aminated lignins may be used to remove dyes and heavy metals from aqueous systems due to their cationic and anionic absorbing capacities (Wang et al. 2014; Xu et al. 2017; An et al. 2020). Several studies related to the synthesis and applications of aminated lignin are listed in Table 1.
Table 1. Reactional Conditions for Lignin Amination and Potential Applications of the Final Products

Perhaps the Mannich reaction is the main way to introduce amine groups in lignin. As a traditional reaction to produce nitrogen-containing compounds, amination may occur in alkaline, neutral, and acidic media. This reaction has been reported with both macromolecular and depolymerized lignin (Ge et al. 2015; Xu et al. 2017; Wang et al. 2018a). The Mannich reaction requires formaldehyde and dialkylaminomethyl groups that react to produce in N,N-dialkylaminomethanol under alkaline conditions (Fig. 4a), and N,N-dialkyl(methylene) ammonium in acid media (Fig. 4b). These amination intermediaries are selective for the H3,5 and G5 positions in lignins and deliver the same reaction product. However, the addition can also occur in phenolates, depending on the reaction pH (Du et al. 2014; Wang et al. 2018a). Figures 4a and 4b show the Mannich reaction for free phenolic OH substructures. However, this reaction can also proceed when lignin is etherified at the C4 position.

Fig. 4. Lignin amination routes based on (a) Mannich in basic media, (b) Mannich under acidic media, (c) use of an amino-silanizating reactant, (d) use of a three-step catalytic synthesis, and e) chlorination followed by amination. Ts stands for the tosyl group.
The Mannich amination has some limitations for S-lignins, which possess less availability of free C5 positions due to their higher methoxylation degree. Besides, Mannich-aminated lignins contain more secondary and tertiary amine groups compared to primary amine groups, limiting further modification of the lignin structure (Pan et al. 2013). Mannich-aminated lignins have excellent potential to be applied as adsorbents for heavy metals and dyes (Liu et al. 2013) and curing agents (Nikafshar et al. 2017), besides value-added applications based on their colloidal properties (Du et al. 2014).
Wang et al. (2018a) improved the reactivity of industrial alkali lignins under acidic conditions using alcoholic depolymerization followed by Mannich reaction with dimethylamine and formaldehyde. Reactions were carried out in an aqueous solution at 60 °C for 4 h under constant stirring with acetic acid as catalyst. These authors were able to anchor 3.40 to 4.17% of N in the lignin structure. Du et al. (2014) used the same conditions to modify industrial softwood kraft lignins using the Mannich reaction, reaching 4.8% N content in the aminated lignins that formed very stable colloidal suspensions in water.
Ge et al. (2015) prepared grafted amine groups in alkali lignin with formaldehyde and methylamine under several conditions. The optimum parameters for achieving 8.32% of N involved the use of 0.16 methylamine/lignin mass ratio and 0.8 formaldehyde/ methylamine molar ratio at 90 °C and pH 12 for 4 h. Aminated lignin was assessed as a biosorbent for lead ions and used as surfactants, polycationic materials, and slow-releasing fertilizers. Liu et al. (2016) prepared a cationic amine emulsifier from a phenolated kraft lignin using the Mannich reaction. Dehydroabietyl and diethylenetriamine groups were used as hydrophobic and hydrophilic moieties, respectively. The resulting material was applied as a cationic asphalt emulsifier in road construction and maintenance due to its foamability, emulsifiability, and surface tension properties.
Another strategy to functionalize lignin with amine groups requires an amine-silanization reagent such as 3-aminopropyl triethoxysilane (APTES). This compound is capable of silanizing and aminating lignin in one step (Fig. 4c). An et al. (2020) reacted a hardwood kraft lignin with APTES under an inert atmosphere and reflux at 70 °C for 24 h. The resulting material containing 6.1% of N was used to remove Congo red and Cu (II) ions from aqueous solutions, showing high adsorption performances by electrostatic attraction and complexation, respectively. APTES reacts with lignin hydroxyl groups, forming an alkoxysilane linkage and exposing a primary amine group at the chain end. The remaining APTES molecules may also react with the first layer, forming a crosslinked APTES multi-layer around lignin. This study deserves attention because it was the first demonstration of a successful amine-silanization of lignin.
Lignin can be aminated using 25% aqueous ammonia and nanoalumina as a catalyst. However, besides the need for catalyst activation, lignin must be demethylated to release free hydroxyl groups that ought to be tosylated prior to amination (Fig. 4d). Nikafshar et al. (2017) obtained aminated lignin with 4.63% of N by this three-step organic synthesis and applied it as a curing agent for epoxy resins. This lignin-based curing agent enhanced the crosslinking density of the epoxy resins and improved their thermal stability (higher glass transition temperature – Tg) and mechanical properties. The authors claimed that this aminated lignin could compete with commercially available curing agents.
Zhang et al. (2011) developed a biobased adsorbent selective to 2,4,6-trinitrotoluene (TNT) from aminated lignin. First, lignin was modified by Friedel-Crafts alkylation followed by amination with a diamine through an SN2 reaction. For alkylation, 1,2-dichloroethane and anhydrous aluminum chloride reacted under reflux at 65 °C and stirring for 6 h to produce chlorinated lignin (ChL). After purification and drying, ChL was solubilized in N,N-dimethylformamide (DMF), and reacted with ethylenediamine at 80 °C for 7 h. Aminated lignin was demonstrated as a suitable adsorbent for TNT at a neutral pH (Fig. 4e).
Lignin can also be aminated after its epoxidation using a diamine. Pan et al. (2013) used propane diamine in different epoxy/diamine molar ratios. Higher diamine loadings favored the obtainment of primary and secondary amines in the lignin structure. Aminated lignins with 5.31 to 6.95% N in their composition were obtained, and these biobased materials were proposed to be used as curing agents or reactant in polyurethane production.
The lignin functionalization with amine groups increases its molecular mass and lowers its Tg. Properties such as reactivity, solubility, and surface tension are increased, tailoring technical lignins for several value-added applications.
Methylation
Methyl groups are very abundant in lignins as part of methoxyl (-O-CH3) groups, and methylation enhances the methoxylation degree of lignins without significantly affecting their molecular mass distribution (Sen et al. 2015; Shen et al. 2020). As a result, methylation increases the solubility in organic solvents and renders lignin more available to specific polymer synthesis applications (Xiong et al. 2020). Lignin methylation was first performed by Brauns (1939) as a method for lignin characterization. Methylation of native spruce lignin was carried out with diazomethane in dioxane and further with dimethyl sulfate in sodium hydroxide. Methylation occurred both in phenolic and enolic hydroxyl groups, rendering lignin insoluble in bisulfite. Today, methylation in dimethyl sulfoxide (DMSO) is one of the most common strategies to modify lignin, making the derivatives less reactive and less thermally stable than the original hydroxylated lignin (Alwadani and Fatehi 2019; Shen et al. 2020; Wang et al. 2015). Furthermore, methylated lignins are more compatible with polymeric materials such as polyethers, polyesters, polyethylene, and natural rubber (Wang et al. 2015) (Table 2).
Table 2. Reactional Conditions for Lignin Methylation and Potential Applications of the Resulting Materials

Methyl groups can be introduced into the lignin structure by nucleophilic aromatic substitution, bimolecular alkyl cleavage nucleophilic substitution, and bimolecular acyl cleavage nucleophilic substitution mechanisms depending on the reactants and reaction conditions (Sadeghifar et al. 2012; Sen et al. 2015). Methylation is more selective for phenolic hydroxyl groups because they are more acidic than aliphatic hydroxyl groups. As a stronger nucleophile, phenolate attacks the methylic carbon atoms in methylating agents, taking the methylic carbon atom for itself (Fig. 5).

Fig. 5. Methylating agents and reactional conditions for the partial, complete, or selective methylation of lignin
Sen et al. (2015) methylated a softwood kraft lignin with dimethyl carbonate (DMC) using a greener protocol compared to other methylation agents considered toxic and hazardous, such as methanol and dimethyl sulfate (DMS). However, the reaction mechanism for DMC methylation under basic conditions was temperature dependent. At 90 °C, DMC acted as a carboxymethylation reagent by a bimolecular acyl cleavage nucleophilic substitution. By contrast, at temperatures above 120 °C, lignin methylation occurred by a bimolecular alkyl cleavage nucleophilic substitution. Therefore, high temperatures were crucial for lignin methylation with DMC. The lignin methylation degree could also be controlled by setting up the reaction time and the DMC to phenolic hydroxyl ratio. The reactional conditions in Fig 5a methylated 100% phenolic OH, 99% carboxylic OH, and 75% aliphatic OH using 10 DMC equivalents to phenolic OH groups (Fig. 5a). However, the use of a base weaker than NaOH at a milder temperature and reaction time led to partial lignin methylation using the same amount of DMC (88.5% phenolic OH, 91% carboxylic OH, and 82% aliphatic OH) (Fig. 5b).
Duval and Avérous (2020) proposed a controlled protocol for lignin methylation using trimethyl phosphate as the methylation agent. Their findings showed that lignin could be partially methylated with precise control and even fully methylated by varying the catalyst amount, temperature, and reaction time. Using 1 equivalent of K2CO3 as the catalyst, trimethyl phosphate acts selectively on phenolic and carboxylic OH, methylating all of them in just 1 h at 120 °C. Otherwise, 20 to 80% of phenolic OH groups were converted when K2CO3 amounts were in the range of 0.125 to 0.5 equivalents. Aliphatic OH groups remained intact under these reaction conditions, ending up as polyols in methylated lignin (Fig. 5c).
Methylation is also a way to protect hydroxyl groups against undesired reactions during lignin conversion. Hence, methylation can be a useful pretreatment strategy for lignin before pyrolysis and thermal conversion. Kim et al. (2019) produced pyrolytic lignin (lignin oil) by solvent liquefaction and noticed that the previous methylation improved the crude oil storage stability in comparison to non-premethylated lignin. In the study, both DMS and DMC were tested as methylating agents. While DMS reacted with lignin at 80 °C for 2 h in 0.7 mol L-1 aqueous NaOH, DMC required more severe reaction conditions of 20 mL g-1 lignin in DMSO plus 0.25 g g-1 NaOH at 130 °C for 15 h. The former methylating condition was selective for phenolic OH, while the latter completely methylated phenolic OH and almost all initial amounts of aliphatic OH (Figd and Fige). Also, pre-methylation increased process yields and decreased char formation after liquefaction. Similarly, Zhu et al. (2016) methylated lignin model compounds prior to a microwave-assisted (400 W) depolymerization in methanol at 100 to 160 °C for 0.5 to 120 min. Pre-methylation facilitated lignin depolymerization, yielding aromatic monomers with reduced oxygen content.
Overall, lignin becomes more hydrophobic upon methylation, with a slightly higher apparent molecular mass and carbon content, lower Tg, higher reactivity, and more extensive intermolecular interactions (Duval and Avérous 2020). Methylated lignin applications are summarized in polymer science as copolymerization components, acting as a source of monomers and intermediates in formulations for lignin-based polymeric materials (Table 2).
Demethylation
Biological, thermo-catalytic, and chemical routes are capable of demethylating lignin, replacing their natural methoxyl groups with hydroxyls, and cleaving aryl-ether linkages in the lignin macromolecule. As a result, more hydroxylated and reactive lignin is achieved with a slightly lower molecular mass distribution. Hydroxyl groups play a primordial role in lignin reactivity because they are easily switchable to new functional groups (Podschun et al. 2017). The conversion of methoxyl into hydroxyl groups renders several potential lignin applications in polymer science to produce polyurethanes, polyesters, and epoxy resins (Table 3).
Table 3. Reactional Conditions for Lignin Demethylation and Potential Applications of the Final Products

Chemical demethylation may be carried out with several reagents and reaction conditions, with sulfur compounds having a great potential to demethylate lignin (Hu et al. 2011). Li et al. (2016) tested several sulfur-demethylating reagents to prepare fast curing agents for phenolic resins. Among them, Na2SO3 produced demethylated lignin in 10 wt% aqueous NaOH at 90 °C for 1 h (Fig. 6).

Fig. 6. Reactional conditions for lignin demethylation according to a) Li et al. (2017), b) Hu et al. (2014), c) Podschun et al. (2017), d) Ma et al. (2021)
Similarly, deprotonated 1-dodecanethiol was used to demethylate pine kraft lignin (Indulin AT – Ingevity, North Charleston, SC, USA) in DMF. The reaction was performed at 130 °C for 1 h under N2 and reflux following an SN2 mechanism. Besides demethylation, partial depolymerization of lignin was evidenced by the lower molecular mass, higher UV absorption spectra, and absence of β-O-4′ ether linkages after demethylation, as demonstrated by proton nuclear magnetic resonance (1H NMR) (Hu et al. 2014).
Podschun et al. (2017) compared conventional and microwave-assisted heating to demethylate organosolv lignin with indium triflate ((In(OTf)3) in water and water/ sulfolane (tetramethylene sulfone, C4H8O2S) mixture. Organosolv lignin was better demethylated under microwave-assisted heating in the presence of the water/sulfolane solvent system. The number of aromatic hydroxyl groups was increased from 2.1 to 4.4 mmol g−1. However, an increase in lignin molecular mass distribution and a decrease in its number of aromatic protons was observed, demonstrating that some level of condensation reactions had occurred. While high molecular mass lignins may help to produce carbon fibers, a great aromatic hydroxyl content opens the prospect for applying lignin as starting material for phenolic and epoxy resins and polyurethanes.
Nikafshar et al. (2017) used Na2SO3 in distilled water to demethylate lignin prior to amination, as aforementioned in the amination section. Chen et al. (2020a) applied Na2SO3 to demethylate corn lignin in aqueous NaOH (33 wt%) for 2 h at 90 °C. The resulting material had its bonding performance increased by oxidation with 20% NaIO4. NaIO4 oxidation produced aldehydes to react with free aromatic sites on demethylated lignin, improving its overall performance as an adhesive for wood-based materials. This chemical demethylation was confirmed by Fourier-transform infrared spectroscopy (FTIR) and by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry. Chen et al. (2020b) demethylated lignin to apply it as polyols for polyurethane synthesis. This functionalization was performed with HBr (48 wt% aqueous) and hexadecyltributylphosphonium bromide (TBHDPB) as a phase-transfer agent in DMF under heating. Demethylation improved the reactivity of lignin and caused a decrease in its apparent molecular mass.
Using a guaiacyl-type structure as an aromatic system to represent lignin, Sawamura et al. (2017) synthesized tannin-like derivatives via demethylation, producing materials with properties and functionality adequate for application as antioxidants, curing agents, adsorbents, and protein precipitation agents. Three substances (1-dodecanethiol, DSH; hydroiodic acid, HI; and iodocyclohexane, ICH) were tested in DMF under reflux for demethylation. HI was the best demethylation reagent compared to DSH and ICH, with the latter leading to recondensation.
Biological demethylation is conducted by fungal (soft-rot, white-rot, and brown-rot) and bacterial enzymes. Among them, bacteria such as Pseudomonas putida, Rhodococcus jostii RHA1, and Sphingomonas sp. SYK6 and fungi such as Penicillium simplicissimum, P. cinnabarinus A-360, Chaetomium piluliferum, and Xerocomus badius are some O-demethylase secreting organisms able to remove the O-methyl groups from lignin, with variable effects on the hydrolysis of aryl ether bonds (Kamimura et al. 2017; Venkatesagowda 2019). By contrast, ligninolytic enzymes such as laccases are known to depolymerize lignin into small fragments besides demethylating it (Kamimura et al. 2019). Venkatesagowda and Dekker (2021) observed that biological demethylation requires long residence times to be effective and that purified O-demethylases are not yet available for industrial applications. Hence, it seems that biological demethylation cannot yet compete with chemical demethylation. For comprehensive reviews on this topic, please refer to Venkatesagowda (2019) and Venkatesagowda and Dekker (2021).
Phenolation
Aromatic condensation (phenolation) in an acid medium modifies lignin by altering its chemical reactivity (Taleb et al. 2020; Thébault et al. 2020). This is because the concentration of phenolic hydroxyl groups is increased, augmenting the number of reactive sites for polymer synthesis and functionalization (Mou et al. 2020). However, for bioprocessing applications, an increment in free phenolic hydroxyl groups may be harmful because these reactive sites can be inhibitory to hydrolases such as cellulases (Mou et al. 2020). Lignin phenolation takes place through different reaction mechanisms (Zhang et al. 2019a). Dehydration in an acidic medium generates reactive sites at Cα and Cγ of lignin side-chains, forming stable benzylic carbocations that may undergo condensation by direct phenol addition. The other routes involve Cγ formaldehyde elimination, enol formation, and addition of phenol in para or ortho positions (Jiang et al. 2018; Londoño Zuluaga et al. 2018). Some of the recent phenolation studies are shown in Table 4.
Table 4. Phenolation of Lignin under Different Conditions

Jiang et al. (2018) conducted a more in-depth study about lignin phenolation with probable structures formed due to this reaction route. According to these authors, apart from breaking most of the ether bonds and secondary reactions, a significant decrease in aliphatic hydroxyl groups was observed due to the phenol incorporation in lignin substructures such as β-O-4′, β-5’/α-O-4′, β-β,’ α-carbonyl, and others. The phenolation process used phenol as a reactant and solvent, and lower acid concentrations (e.g., 5%) were used to improve the viability of the production process.
Taleb et al. (2020) studied both phenolation and acetylation of spent coffee ground lignin after pretreatment with dilute sulfuric acid. Compared to acetylation, phenolation led to more thermally stable (430 °C) lignin streams, having a higher availability of OH sites. Moreover, phenolated lignin had a superior adsorption performance of methylene blue (recovery of 99.6%), a compound used to evaluate the cationic adsorbent capacity of dyes. This study demonstrated a different application for lignin and offered a low-cost material to treat textile effluents. Although relatively inexpensive, the recovery and reuse of phenolated lignin were not demonstrated in this study.
Recently, phenolated Eucalyptus sp. alkaline-extracted lignin was studied as a matrix for cellulase adsorption. Lignoresorcinol (LigR) and lignopyrogallol (LigP) phenolated lignins (Fig. 7) had maximum cellulase adsorption capacities of 842.1 and 911.4 mg g-1, respectively, compared to 76.5 mg g-1 of the starting material. The enzymes were removed from both phenolated lignins by changing the pH from 10 to 4.8, with LigP adsorbing ten times more cellulases than lignin without modification (Mou et al. 2020). Compared to LigR, LigP provided a better enzyme migration to fresh cellulosic materials during the enzymatic hydrolysis stage, with LigP-desorbed enzymes displaying a higher total cellulase activity and a better hydrolysis performance.

Fig. 7. Structure of pyrogallol and resorcinol phenolated lignins
Thébault et al. (2020) evaluated the effects of lignin type and substitution degree on the properties of phenol-formaldehyde resins based on phenolated lignins. A factorial design was carried out using kraft and lignosulfonate lignins, unmodified and phenolated lignins, and level of phenolation (30 or 50%) as independent variables. Phenolation increased the number of reactive sites and decreased the average molecular mass of lignin. As a result, phenol-formaldehyde resins based on phenolated lignins had surface tension, viscosity, molecular mass distribution, and reactivity higher than those derived from unmodified lignins.
Wang et al. (2020) synthesized lignin-containing phenol-formaldehyde wood adhesives (LPF) from fractions of an industrial birch alkaline lignin that was previously submitted to sequential solvent extraction with isopropyl alcohol (i-PrOH), ethanol (EtOH), and methanol (MeOH). All these fractions were characterized to elucidate LPF structure/performance correlations. Carbohydrate, ash, and Klason lignin contents increased along this solvent extraction sequence and lignin apparent weight-average molecular mass (Mw). Lignin, phenol, and formaldehyde were used as reactants using a one-pot reaction system to produce LPF. Lignin was integrated covalently into the phenol-formaldehyde resin, and this was correlated to its adhesive strength. Also, the incorporation of lignin with high Mw and a high degree of condensation (MeOH lignin) affected the resin adhesiveness. This work was the first to demonstrate the feasibility of fractionating industrial birch alkaline technical lignins to produce thermoset LPF wood adhesives.
Depolymerization followed by phenolation was carried out by Zhou et al. (2020) to synthesize lignin-based phenolic foams. Under optimal reaction conditions, both Mw and number-average molecular mass (Mn) decreased from 12.600 and 6.480 g mol-1 in alkaline lignin (AL) to 6.100 and 1.500 g mol-1 in phenolated alkaline lignin (PAL). In addition, lignin phenolic hydroxyl groups increased from 2.4 in AL to 3.3 mmol g-1 in PAL. Foams were also characterized for their physical, mechanical, thermal, and morphological properties. Both macro and micro images of PAL-based and AL-based phenolic foams revealed that the foam structure remained uniform for lignin incorporation up to 30%. Both samples had better thermal stability, lower volumetric water absorption, and lower slag rate than foams synthesized in the absence of lignin. Also, for materials with the same degree of substitution, PAL-based phenolic foams had higher compressive strength and a more uniform structure than AL-based foams.
Sulfomethylation
Low water solubility is a limiting factor for the valorization of kraft lignins. In this context, lignin sulfomethylation produces water-soluble derivatives by introducing a methylene sulfonate group in aromatic rings (Aro and Fatehi 2017). This process differs from sulfite pulping, whereby sulfonic acid groups are placed in the lignin aliphatic side-chains. Figure 8 shows that lignin sulfomethylation occurs by adding sodium sulfite anions into alkaline media, preferably at the unsubstituted C5 of lignin substructures. This reaction takes place at various pH (7 to 13), temperatures (60 to 160 °C), reaction times (0.5 to 9 h), and sulfite/lignin and formaldehyde/lignin mass ratios of 0.1 to 1.0 and 0.01 to 1.0, respectively (Aro and Fatehi 2017; Eraghi Kazzaz et al. 2019; Konduri and Fatehi 2015). He and Fatehi (2015) studied the sulfomethylation of LignoForceTM kraft lignins using formaldehyde (HCHO) and Na2S2O5. The maximum estimated sulfonation degree was achieved at 97.1 °C for 3.2 h using 0.97/1 HCHO-to-lignin and 0.48/1 Na2S2O5-to-lignin molar ratios.
Qin et al. (2015) investigated the use of grafted sulfonated alkali lignin (GSAL) as a dispersant for coal-water slurries. GSAL was synthesized by sulfomethylation followed by etherification and polycondensation to obtain high levels of sulfomethylation and high molecular mass distributions. In this application, sulfomethylation was carried out for 1 h at pH 10 and 60°C.

Fig. 8. Scheme of sulfomethylation of lignin with sodium sulfite (adapted from Eraghi Kazzaz et al. 2019)
Different conditions in lignin sulfomethylation allow the obtainment of sulfomethyl-derivatives with various degrees of substitution, leading to materials with different average molecular masses and degrees of sulfonation. This reaction has been applied to kraft lignins by the MeadWestvaco Corporation (now Ingevity) to produce dye dispersants marketed as Reax® since the 70’s (Meister 2002). Sulfomethylated lignins (SML) have also been used as water reducer for cement admixtures (Kamoun et al. 2003), flocculant in water purification systems (Bolto and Gregory 2007), dispersant in pesticide formulations (Li and Ge 2011), and also as potential corrosion inhibitors for iron-based materials (Abu-Dalo et al. 2013).
Sulfomethylation side reactions include sodium thiosulfate formation, which may be avoided using high temperatures (100 to 150 °C) and sodium sulfite in excess to improve SML yields. Another hindrance is found in the lower reactivity of hardwood lignins because, unlike guaiacyl, the nucleophilic attack of phenolic hydroxyl groups is hindered by its high degree of methoxylation.
Konduri and Fatehi (2015) studied the sulfomethylation of hardwood kraft lignins. The main objective of the work was to produce water-soluble kraft lignin with an anionic charge. The optimal reaction conditions involved a lignin concentration of 20 g L-1, 0.5 mol L-1 of NaOH(aq), 0.9 sodium hydroxymethyl sulfonate/lignin molar ratio, 100 °C, and 3 h of reaction time. These conditions provided an SML with a charge density of −1.6 meq g-1 and 1.48 mmol g-1 sulfonate groups, while the unmodified lignin had a negligible charge density and 0.03 mmol g-1 sulfonate groups. The SML solubility in water at neutral pH was successfully improved to 40 g L-1, in contrast to the insolubility of the unmodified lignin. Further increments in the degree of sulfomethylation were not possible due to the formation of undesirable byproducts such as sodium thiosulfate. However, the density of sulfonate groups achieved in this study was higher than that obtained by Wu et al. (2012) after sulfomethylation of corn stalk alkaline lignin, reaching 1.29 mmol g-1 as reported in the literature.
Huang et al. (2018) performed the sulfomethylation of alkaline lignin (AL), and enzymatic hydrolysis lignin (EHL) derived from alkali-pretreated bamboo fibers. SML yields of circa 95% were achieved from AL sulfomethylation after 3 h at 110 °C using a sodium hydroxymethylsulfonate/lignin molar ratio of 1.0. The maximum lignosulfonate yield from EHL was only 68.9% when the reaction was carried out for 4 h at 110 °C with a sodium hydroxymethylsulfonate/lignin molar ratio of 0.8. The largest sulfomethylation of AL was attributed to the availability of more reaction sites (free C5 position of lignin), possibly due to its lower average molecular mass and the presence of residual carbohydrates in EHL.
One additional drawback of sulfomethylation is the use of formaldehyde, which is carcinogenic, mutagenic, and environmentally unfriendly. Hence, recent studies are focused on developing alternative sulfoalkylation reaction routes to alleviate its environmental impact. For instance, lignin sulfobutylation provides water-soluble lignin derivatives similar to conventional sulfomethylation with the advantage of being carried out in aqueous media (Eraghi Kazzaz et al. 2019; Hopa and Fatehi 2020; Huang et al. 2018).

Fig. 9. Sulfobutylation of phenolic and aliphatic hydroxyl groups in lignin with 1,4–butane sultone (adapted from Hopa and Fatehi 2020)
Sulfobutylation is a one-step reaction in which 1,4–butane sultone reacts with both phenolic and aliphatic hydroxyl groups of lignin (Fig. 9). In general, sulfobutylated lignin (SBL) is produced at 70 °C for 6 to 7 h at pH 12. Although sulfobutylation requires temperature and pressure lower than sulfomethylation, the sulfobutylation reagent is more expensive. Also, sulfobutylation leads to a higher degree of substitution because sulfomethylation is limited to lignin phenolic hydroxyl groups (Eraghi Kazzaz et al. 2019; Hopa and Fatehi 2020).
Hopa and Fatehi (2020) compared both sulfobutylation and sulfomethylation of a softwood kraft lignin to produce sulfoalkylated lignins with similar charge densities (circa -2.3 meq g-1). Both products were effective as kaolin dispersants at neutral pH. One advantage of sulfobutylation was that the quantity of reagents to achieve similar charge densities was lower than that of sulfomethylation. The required 1,4-butane sultone/lignin molar ratio for SBL was 0.2, while an equivalent charge density in SML was only achieved using a formaldehyde/lignin molar ratio of 2 and a sodium metabisulfite/lignin molar ratio of 1. These findings showed the possibility of obtaining sulfoalkylated lignin derivatives with similar properties under more environmentally friendly conditions.
Oxyalkylation
Lignin has a high potential to react through oxyalkylation with reagents such as ethylene oxide, propylene oxide, and others (Cateto et al. 2009; Wu and Glasser 1984). Particularly, oxyalkylation has been recognized for overcoming lignin’s poor solubility and its frequently observed adverse effects on mechanical properties of solid polymers, viscosity, and cure rate of resin systems (Wu and Glasser 1984). Chemical modification by oxyalkylation has been demonstrated to offer a route to improve solubility, to reduce the brittleness of lignin-derived polymers, and to upgrade viscoelastic properties in various uses, such as prepolymer for engineering plastics (Li and Ragauskas 2012), for polyurethane films (Saraf and Glasser 1984), and rigid polyurethane foams (Zhang et al. 2019b). Also, oxyalkylation shows that all phenolic hydroxyl groups can fully be converted into lignin-polyether chains with hydroxyl groups, increasing the reactivity by reducing the sterically hindered phenolic OHs (Kühnel et al. 2017; Zhang et al. 2019b).
Lignin oxyalkylation may result in a copolymer combining covalently high modulus lignin with a lower modulus aliphatic polyether phase. In the industry of polymers, this reaction is a comprehensive method for the production of polyols as precursors for the polyurethane production, especially with propylene oxide (PO), since the procedure provides a chain extension reaction forming grafts of poly(propylene oxide) (Kühnel et al. 2017; Zhang et al. 2019b).
The oxyalkylation of lignin with epoxides enables the substitution of conventional petrochemical polyols by a renewable polyol source (lignin) and produces interesting macromonomers for polyurethane synthesis, namely bio polyols. This is interesting since polyurethanes are used in many industries due to their wide-ranging mechanical properties and their ability to be relatively quickly processed in various forms (flexible and rigid foams, elastomers, adhesives, etc.) (Berrima et al. 2016; Kühnel et al. 2017).
The reaction of lignin with PO is an etherification that requires a catalyst, commonly alkaline (KOH), but also acidic catalysts such as H3BO3, Al2O3, AlCl3, ZnO can be employed (Fig. 10). The reaction is usually carried out at high temperatures (>150 °C) and high pressure (>10 bar). The resulting products from oxypropylation are a mixture of oxyalkylated-lignin and poly (propylene oxide) (PPO) diols, and the last arises from the homopolymerization of PO. Both materials were already applied to synthesize rigid polyurethane foams (Wu and Glasser 1984; Lee et al. 2017; Zhang et al. 2019b).

Fig. 10. Scheme of oxypropylation of lignin for bio polyol production; adapted from Zhang et al. (2019b)
The pioneering investigation on oxypropylation of lignins was carried out by Wu and Glasser (1984). PO was employed in alkaline conditions (KOH) at 180 °C for the hydroxypropylation of lignin and lignin-like model compounds. It was observed that both homopolymerization and copolymerization occurred during batchwise hydroxyl-propylation of lignin-like model compounds and lignin. There was a dependence between the increase of reaction rate and the increasing KOH concentration until 2.6 mmol mol-1 PO. Also, the retardation effect of functionalization on the initial rate of PO homopolymerization decreased in the order of COOH > phenolic OH > aliphatic OH. Interestingly, the authors showed that the volume of the solvent used in the reaction could be as low as 1 to 1.5 mL PO g-1, which is advantageous, since the solvent used was toluene, a toxic, flammable, and volatile compound. Several of these findings, such as the effect of catalyst concentration on reaction rates, optimal temperature, and product viscosity, were used in subsequent investigations regarding oxypropylation.
Lee et al. (2017) performed the oxypropylation of methanol-insoluble softwood kraft lignin at 40 °C and 1 atm for 12 h in the presence of NaOH. Afterward, the modified kraft lignin was reacted with sebacic acid or polybutadiene (dicarboxy terminated) to produce monomers for polyester synthesis. The use of mild reaction conditions in the absence of organic solvents was claimed beneficial for the process economics and for the environment. The polyesters prepared in this way presented a higher decomposition temperature compared to other lignin-based polyesters. The temperatures were enhanced from 217 °C and 367 °C (oxypropylated lignin) to 380 °C and 453 °C for polyesters derived from sebacic acid and polybutadiene, respectively. Despite these advantages, the synthesis of polyols with propylene oxide is discouraged because PO is a harmful chemical due to its high vapor pressure, explosiveness, flammability, toxicity, and carcinogenicity (Kühnel et al. 2017).

Fig. 11. Scheme of oxyalkylation of lignin with cyclic organic carbonates via (a) transesterification and (b) etherification (adapted from Kühnel et al. 2017)
Recent studies have focused on the substitution of PO by safer and non-toxic reactants, such as cyclic carbonates (Kühnel et al. 2017; Zhang et al. 2019b). The oxyalkylation with cyclic carbonates occurs with the deprotonation of OH groups followed by the nucleophilic attack at either carbonyl (transesterification) or alkylene (etherification) atoms of the carbonate (Fig. 11). Due to the reversibility of transesterification, etherification is favored during oxyalkylation. The catalysts used in these cases usually are alkaline (K2CO3, LiCO3, NaOH, or KOH).
Kühnel et al. (2017) studied the use of various cyclic organic carbonates in the oxyalkylation of lignins from different sources (hardwood, softwood, annual plants) and pulping processes (organosolv, kraft, soda). The alkylation reactants used were ethylene (EC), propylene (PC), butylene (BC), and glycerol carbonate (GC). The alkali catalyst used was 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), and the reaction was performed at 170 °C in N2 atmosphere for 3 h. For all lignins, higher degrees of substitution (DS) were achieved using cyclic organic carbonates in the following order EC > GC > PC > BC. The authors showed that impurities (ash and sulfur) and the molecular mass of the lignins have no significant influence on the degree of oxyalkylation. Notably, despite high temperatures (170 °C), an almost total OH substitution could be reached (DS = 0.96) for hardwood kraft lignins.
Zhang et al. (2019b) developed oxyalkylated lignin from the reaction of kraft lignin (KL) with ethylene carbonate (EC) and polyethylene glycol (PEG400). EC was the oxyalkylation agent, and PEG400 with water was co-solvent and additive to tailor the viscosity and OH content of final polymers obtained from the reaction of oxyalkylated lignin with isocyanate. The optimum conditions to oxyalkylate the KL were 170 °C, reaction time of 30 min, PEG400/EC mass ratios of 50/50 to 70/30, and KL mass loadings of 20 to 50 wt%. More than 80% of OH content in the product was aliphatic, being more accessible, which resulted in an increase of 14-fold in the reactivity for the subsequent reaction (oxyalkylated KL with isocyanate) in comparison to that for the unmodified KL. This study showed a new strategy for obtaining tailored rigid polyurethane foam from lignin that showed the matter of the optimization of the lignin oxyalkylation.
Esterification
Esterification is one of the main strategies for the functionalization of primary and secondary OH in lignin. Ordinarily, lignin esters are obtained using several anhydrides and acid chlorides in a pyridine or dioxane medium (Liu et al. 2019).
Since Fischer reacted carboxylic acids with alcohols at high temperatures for the first time, several modifications have been made in this route to achieve high yields from a vast range of reagents and raw materials. However, the Fischer route can be problematic for lignin because it contains several reactive sites and may undergo structural modifications when exposed to acids and heat (Liu et al. 2019). Besides, partial esterification often occurs, depending on the reaction conditions (Luo et al. 2018). Esterified lignin has altered properties such as higher solubility in non-polar organic solvents, lower melting point and enhanced thermal and oxidation stabilities. As a result, lignin is made more compatible with hydrophobic matrices such as polyolefins and aliphatic polyesters (Buono et al. 2016; Sameni et al. 2017; Shayesteh et al. 2020). One drawback of most lignin esterification methods is that toxic solvents are applied, and hazardous wastes (halogenated species) are generated, particularly for homogeneous catalytic systems (Liu et al. 2019). A summary of recent studies regarding lignin esterification is found in Table 5.
Table 5. Lignin Esterification Routes and Some Applications

Acetylation is a classic method for esterification that is generally performed with acetic anhydride or acetyl chloride using pyridine as a catalyst. However, the use of acetic acid as the acylating agent is encouraged because it is relatively cheap, sustainable, and widely available in the market (Shayesteh et al. 2020).
De Oliveira et al. (2020) studied the acetylation of kraft lignin using acetic acid as both solvent and acetylating agent. Reactions were carried out under microwave heating, and products were characterized by FTIR, NMR (1H, 13C, and 31P), and heteronuclear single quantum correlation (HSQC). The resulting lignin acetates had excellent solubility in organic solvents (chloroform, ethyl acetate, and dichloromethane) compared to unmodified kraft lignin. Aliphatic OHs were selectively acetylated by this method. Consequently, the antioxidant capacity was also improved compared to commercial antioxidants such as butylated hydroxytoluene (BHT), paving the road for its utilization as a sustainable green antioxidant additive.
The use of acetylated lignin in poly(lactic acid) (PLA) composites has also been described in the literature. Yetiş et al. (2020) isolated a high lignin content microfibrillated cellulose and modified its surface by acetylation with acetic anhydride to apply it as a reinforcement agent for PLA. Although base-catalyzed, the reaction had no effect on the crystalline structure of microfibrillated cellulose, resulting in lignin-containing PLA composites with improved mechanical properties and thermal stability.
Acetylated lignins were applied to produce polyacrylonitrile fibers (PAN) without releasing toxic byproducts such as hydrogen cyanide (HCN). Zhang and Ogale (2014) acetylated a softwood kraft lignin that was further dry-spun into precursor fibers and processed into carbon fibers with low diameters (~7 mm) and adequate tensile strength. Acetylation was carried out using a low anhydride concentration (0.66 mL Ac2O g-1 lignin) for only 0.25 h. Significant portions of lignin hydroxyl groups were left unreacted to facilitate crosslinking during the subsequent processing steps.
Li et al. (2020) used acetylation as a pretreatment technique for lignin pyrolysis. Acetylation prevented the generation of quinone intermediates from hydroxyls that could produce pyrolytic coke, which is known to decrease bio-oil yields. Also, pyridine was replaced by a non-toxic quaternary ammonium salt, choline chloride (ChCl). The reaction was carried out with 1 g of pine kraft lignin (PKL) plus 6 g of ChCl/acetic anhydride, which were heated up to an unspecified reaction temperature. The bio-oil yield increased from 34.9% to 40.9% when the acetylation time was increased from 3 to 60 min.
Esterifying agents other than acetic acid and acetic anhydride have also been used for lignin esterification. He et al. (2018) used citric acid to modify cellulose nanocrystals and lignin nanoparticles (LNP). Freeze-dried LNP were mixed with 5 wt% citric acid and 1 wt% sodium hypophosphite for 1 h. Then, the mixture was transferred to a vacuum oven for 14 h to produce a modified LNP with newly formed ether and ester bonds (MLNP). The total mass of MLNP was 17% higher than that of the starting LNP. FTIR and NMR analyses confirmed the grafting of methylene groups derived from citric acid (R2CH2) and also the presence of ester carbonyl (C=O) groups. In comparison with LNP, MLNP presented higher thermal stability due to the formation of aryl-ether linkages and increased hydrophobicity, which reduced its dispersion in methanol. This study suggested that esterified and etherified lignin could be readily dispersed into the hydrophilic polymers such as polyvinyl alcohol and used as nanofillers.
Indulin AT (pine kraft lignin) and Protobind lignin (derived from soda pulping of agricultural residues) were esterified by Luo et al. (2018). Reactions were performed with butyric and crotonic anhydrides with 1-methylimidazole (1-MIM) as the catalyst at 120 °C for 24 h. Extensive lignin esterification was demonstrated by the appearance of ester carbonyl groups (C=O) in its FTIR spectrum with the concomitant disappearance of O-H vibrational modes. Further, the modified lignin was coated with dicumyl peroxide (DCP) followed by hot pressing at 175 °C and 5 MPa for 10 min. Then, these three lignin-based materials (esterified, esterified, and thermally-treated, and esterified, thermally-treated and DCP-coated) were assessed by differential scanning calorimetry (DSC) analysis and Thermal mechanical analysis (TMA). By far, the DCP-coated crotonylated lignin had the highest Tg, whereas the Tg of DCP-coated butyrated lignin remained practically unaltered. This is because, by contrast to butyration, crotonylation introduces a double bond in the lignin structure, improving its susceptibility to radical polymerization and leading to highly crosslinked DCP-coated lignin esters.
Nasiri et al. (2020) synthesized pressure-sensitive adhesive films incorporating water-soluble (HPH) and water-insoluble (LPH) kraft lignins. HPH was esterified by maleation and LPH by acetylation and acrylation. These modifications were carried out to improve lignin miscibility with synthetic monomers such as butyl acrylate (BA) and methyl methacrylate (MMA) for latex production. The incorporation of 10 wt% maleated lignin in latex improved tack, peel strength, and shear strength of the acrylic adhesive formulation, while 5 wt% of acrylated lignin increased tack and shear strength. By contrast, the incorporation of acetylated lignin did not produce stable compounds. Both maleated and acrylated lignins increased the particle size and lowed the latex viscosity. Nevertheless, their incorporation into the polymer matrix was claimed as feasible and promising.
Ko et al. (2020) prepared a blend of 50 wt% polyvinyl alcohol-lignin (PVA-lignin), resulting in a resin with high strength, stiffness, and adhesion performance. Further, this PVA-resin was esterified at 180 °C with 40% maleic anhydride (MA). Thermogravimetric analysis revealed excellent thermal stability, which was attributed to the covalent bonds formed in the esterified material. Moreover, good waterproof and adhesive performances were observed, while tensile strength, toughness, and failure strength were increased by 18, 31, and 55% concerning the pristine PVA-lignin resin. This environment-friendly resin was suggested as a strong candidate for natural fiber reinforced polymer composites.
Epoxidation
Epoxy materials are used in many civil engineering applications and the automotive industry, being ideal for developing metallic adhesives (Xiang and Xiao 2020).
Table 6. Main Lignin Epoxidation Routes and their Applications

Many epoxy materials are available in the market, but the most widely used is the diglycidyl ether of bisphenol A, which has been shown to be highly toxic (Marqueño et al. 2019). For this reason, several research groups have tried to use lignin as an environmentally friendly, renewable substitute for bisphenol A (Table 6).
Three main approaches can be used to epoxidize lignin: (1) direct epoxidation; (2) chemical modification prior to epoxidation; or (3) direct incorporation into epoxidated polymers (Gouveia et al. 2020). The first approach does not result in high epoxy contents due to the poor solubility and low reactivity of lignin. The second approach was designed to introduce more reactive functional groups in lignin for epoxidation by methods such as demethylation, phenolation, and hydroxymethylation. Unfortunately, an increase in lignin reactivity tends to compromise the mechanical properties of lignin-based epoxy resin compared to raw lignin (Gouveia et al. 2020). Since the third approach is dedicated to incorporating lignin into epoxidated polymers, it will not be described in this review because it does not involve any direct lignin modification.
The most common method to epoxidate lignin uses epichlorohydrin in warm basic media. Hydroxyl groups are ionized or deprotonated in a strong basic media to form an alkoxide anion. In this respect, phenolic lignin groups are easily ionized at a pH above 9, while aliphatic hydroxyls require a pH above 12 for the same (ETB, 2021). Alkoxide groups promote a nucleophilic attack on the carbon atom in epichlorohydrin, opening the oxirane ring and forming chloroglyceryl ether bonds with lignin (Jablonskis et al. 2018). Afterward, the oxirane rings are restored under alkaline conditions, releasing KCl and creating a strong reaction site for anchoring new functionalities in the lignin structure.
Chen et al. (2016) synthesized emulsifiers from lignin by epoxidation and etherification. First, epoxidation of alkali lignin was performed using epichlorohydrin in an aqueous medium containing tetrabutylammonium bromide at 90 °C for 5 h with constant stirring. After this, a NaOH solution was added, and the reaction went on for another 5 h at 90 °C to close the oxirane ring. After several washing steps with acetic acid, distilled water, and anhydrous sodium sulfate, the product was shown to contain 0.32 mol of epoxy groups per 100 g of modified lignin. Etherification of epoxidized lignin with polyethylene glycol was carried out in an oil bath at 130 to 140 °C for 5 h using potassium peroxydisulfate as a catalyst. Stable emulsions were achieved by adding 1.03 wt% of the modified lignin in water, reaching a surface tension of 43.3 mN m-1.
Liu et al. (2013) synthesized aminated epoxy-lignins for the adsorption of heavy metal ions (Cu, Pb) in water. Purified kraft lignin from black liquor was epoxidated in NaOH containing epichlorohydrin for 4 h at 90 °C. Then, amination was achieved in NaOH using diethylenetriamine and formaldehyde. The products were obtained by filtering, washing with distilled water, and drying at 70 °C. FTIR confirmed epoxidation and amination, but the extent of lignin modification was not quantified. The synthetic material exhibited high adsorption levels for Cu (~67 mg g-1) and Pb (~50 mg g-1).
Yang et al. (2014) modified lignin by hydromethylation, epoxidation, and phenolation to apply them to synthesize polyurethane foams (PUF). PUF was produced on a laboratory scale with polyether glycol, blowing agent, and catalysts mixed with 4,4-methylene diphenyl diisocyanate at a known mass ratio and complete mixing. The highest content of phenolic hydroxyl groups (8.49%) in hydroxymethylated lignin was attained at 70°C, 4 h, and 1 wt% of unmodified lignin. For both epoxidation and phenolation, phenolic hydroxyl contents of 6.21% and 9.41% were achieved at 80 °C for 4 h and 95 °C for 3 h, respectively. Among all PUF produced (phenolated lignin-based PUF, epoxidated lignin-based PUF, hydroxymethylated lignin-based PUF, and neat PUF), lignin-based synthetic PUF had slightly higher thermal stability and compression strength with the addition of 1% of phenolated lignin. This suggests that high phenolic hydroxyl contents in lignin play an essential role in developing these properties.
Cortés-Triviño et al. (2018) grafted lignin with poly(ethylene glycol) diglycidyl ether (PEGDE) for application as a thickener in bio-lubricant formulations. The epoxidation process was carried out by suspending kraft lignin (10 g) in 20 wt% NaOH with different amounts of PEGDE at 30 °C for 3 h. The product was obtained by filtration and washing with distilled water. The highest epoxy index of 0.79 mol kg-1 was obtained using the lowest quantity of PEGDE (2.5 g). The epoxy-lignin was then mixed with castor oil to obtain an oleogel. The viscosity, consistency, and viscoelastic properties increased with the epoxy index of the modified lignin.
Phosphorylation
Phosphorylated materials have been applied in flammable plastics as flame retardants due to their high thermal stability. Renewable flame retardants can be produced from lignin by phosphorylation of phenolic and aliphatic hydroxyl groups, opening a real market opportunity for its valorization. Also, the thermal decomposition of lignin releases char by rearrangement of unstable radicals.
Yu et al. (2012) used wheat straw kraft lignin to produce flame retardants for polypropylene by grafting phosphorus and nitrogen in its structure. Phosphorylation was performed in DMF containing imidazole, Et3N, and POCl3 as the phosphorus donor. The reaction was carried out at 95 °C for 12 h, and purification was performed by washing with methanol, yielding 92% of phosphorylated lignin with an 8.1 wt% phosphorus content. Compared to kraft lignin, phosphorylation led to an increase of 20% in char formation at 600 °C under the N2 atmosphere. Incorporating the modified lignin into polypropylene reduced its heat release rate and slowed its combustion process, demonstrating a good performance as a flame retardant. Table 7 summarizes the main lignin phosphorylation routes that are found in the literature.
Table 7. Main Lignin Phosphorylation Routes According to the Literature

Prieur et al. (2016) phosphorylated kraft lignin to be added as a flame retardant of acrylonitrile butadiene styrene (ABS). Phosphorus grafting was achieved by dissolving lignin in THF with the addition of P2O5. The reaction was performed under reflux for 7 to 8 h, by which time phosphorylated lignin was largely precipitated. Water was added to inactivate the phosphorylating agent, and THF was removed by evaporation, resulting in the incorporation of 4.0 wt% of phosphorus in lignin. Incorporation of 30 wt% of this modified lignin into ABS resulted in composite with a significant reduction of its heat release rate. Also, phosphorylation increased the thermal stability of kraft lignin by 20 wt%. The authors claimed that this reaction system could be scaled up for future industrialization.
Gao et al. (2020) phosphorylated lignin in an aqueous medium containing urea and ammonium dihydrogen phosphate as the phosphorylating reagent. The mixture was reacted at 70 °C for 60 min with constant stirring. Next, lignin was dried at 70 °C and cured at 150 °C for 4 h. The loosely attached chemicals after the curing step were removed using cold deionized water. Phosphorylation by nucleophilic substitution of phenolic and aliphatic hydroxyl groups was confirmed by 1H NMR, 31P NMR, and X-ray photoelectron spectroscopy (XPS). The resulting material exhibited a maximum decomposition temperature of 620 °C compared to 541 °C of the unmodified lignin.
Matsushita et al. (2017) synthesized an advanced biobased flame retardant from kraft lignin by introducing an intumescent group (3-amino-1,2,4-triazole) into the lignin structure followed by phosphorylation with phosphoryl chloride for 1 h at room temperature. Afterward, phosphoryl end groups were crosslinked using several alkane diols in pyridine, with the final products being purified by washing with water and methanol. Phosphorous incorporation was relatively high in the range of 6 to 10%, and this was directly related to the quantity of phosphoryl chloride and the chain length of alkane diol used. In simple combustion tests using a gas burner at 1300 °C, the resins exhibited non-burnable properties, producing only a swollen char. Also, the peak heat release rate was significantly lower than that of a commercial phenol-formaldehyde resin.
Costes et al. (2016) performed a two-step phosphorus/nitrogen grafting of kraft and organosolv lignins to be used as a flame retardant additive in poly(lactic acid) (PLA). For the first step, lignin was solubilized in chloroform and heated to 50 °C, then POCl3 was added dropwise. The reaction continued for 17 h with constant stirring at 60 °C. The product was recovered by filtration and washed with fresh chloroform to remove the POCl3 excess. Lignin was dried at 60 °C in a vacuum oven. In the second step, nitrogen was added by treating the modified lignin with aqueous NH4OH at room temperature for 2 h. The final products were obtained by washing, filtration, and drying. Both kraft and organosolv modified lignins incorporated about 9% of phosphorous and 7% of nitrogen. The addition of 20 wt% of these lignins to PLA reduced its heat release during combustion owing to the formation of an insulating char layer at the surface of the burning sample.
Nada and Hassan (2003) phosphorylated cotton stalks kraft lignin and applied it as an adsorbent for heavy metal ions. Cotton stalks, anthraquinone brown stocks, and bleached pulps were also used for comparison. Phosphorus was grafted in all fractions using phosphorus oxychloride as the phosphorylation agent. Reactions were carried out at 115 °C for 2 h in a pyridine and dichloromethane mixture, and products were obtained after filtration, washing with dilute HCl (0.1 mol L-1), and distilled water, and drying. Lignin fraction was shown to incorporate a phosphorus content (33.5 mg g-1) higher than that of the other cellulosic fractions. All modified materials exhibited high adsorption capacities for Cr and Cu ions (~1 mg g-1). The authors claimed that phosphorylated lignin had the highest adsorption capacity, but the results do not appear to have been statistically significant.
Nitration
Lignin nitration is a simple process that is usually performed using non-aqueous solvents. The most widely used nitrating agents are nitric acid in concentrated acetic acid, acetic anhydride, or sulfuric acid (Meister 2002). The resulting nitrolignin (NL) is an amorphous powder, ranging in color from yellow to brown with an average molecular mass between 600 to 2000 Da. Nitration causes an extensive lignin dealkylation, and nitrogen incorporation is generally around 7 wt% (Zhang and Huang 2001; Meister 2002).
Lakhmanov et al. (2020) performed the nitration of lignin with nitric acid in binary water-aprotic solvent mixtures. Acetyl nitrate, a mixed anhydride of nitric and acetic acids, was also used as the nitrating agent. When nitration was performed with nitric acid, the maximum yield of nitrated lignin (83 to 101%) was achieved using 1,4-dioxane, acetonitrile, and tetrahydrofuran, while the maximum nitrogen content (4.3 to 4.5%) was obtained using 1,4-dioxane or acetonitrile. The use of DMSO and DMF showed product yields of 23 to 35% and nitrogen contents of 1.3 to 3.9%, but their high oxygen contents were indicative of depolymerization and some oxidative transformations. The nitration with acetyl nitrate, on the other hand, formed the product with up to 4.7% nitrogen content.
NL has been identified as a promising candidate for electrochemical applications. For example, nitrogen-doped carbons derived from NL were used for the electrochemical reduction of oxygen, showing catalytic activities comparable to non-noble metal catalysts (Graglia et al. 2016). NL was used together with a castor oil-based polyurethane (PU) prepolymer to produce a polymer network. Covalent bonds were formed between the PU prepolymer and NL, forming a crosslinked material with good mechanical properties and high thermal stability. Also, NL showed a thermal reactivity with PU much higher than nitrocellulose (Zhang and Huang 2001).
Sulfonation
Sulfonation is a reaction whereby sulfonate groups are added to lignin using sulfuric acid or sodium sulfite as reagents (Meister 2002). The increase in the sulfur content in lignin correlates with an increase in its dispersion performance in suspensions (Lou et al. 2013). The kraft lignin sulfonation occurs in the α-position of phenylpropanoid units, which is commonly occupied by α-O-4′ aryl-ether and other ether bonds. Hence, the complete sulfonation of lignin is impaired by such ether bonds in its structure (Chakar and Ragauskas 2004; Crestini and Argyropoulos 2002).
Functionalization such as demethylation, phenolation, hydroxymethylation, reduction, oxidation, or hydrolysis increase kraft lignin reactivity toward sulfonation, yielding highly charged sulfonated lignins (Filley et al. 2002; Hu et al. 2011; Meister 2002). Among these, phenolation is more effective than reduction, oxidation, and hydrolysis to improve lignin reactivity (Alonso et al. 2005).
Kraft lignin can be sulfonated directly by reacting with >95% sulfuric acid at temperatures below 40 °C (Dilling 1991). Then, sodium carbonate is used for neutralization, precipitating the sulfonated lignin (SL) as its sodium salt. This process was reported to increase the presence of sulfonate groups kraft lignin by 2.5 mol mol-1. However, the downstream processing for SL purification can be complicated due to its high solubility in water. Membrane filtration is an option for this purpose at the expense of a significant increase in operating costs.
Softwood kraft lignin was modified by phenolation followed by sulfonation with sulfuric acid or sodium sulfite to produce water-soluble lignin materials. Pretreatment by phenolation generated SL preparations with a high charge density of 3.12 meq g-1 and high solubility in aqueous systems. Treatment of phenolated lignin with sodium sulfite generated sulfonated lignins with a lower charge density of 1.20 meq g-1, mostly due to the partial etherification of the most reactive α-position. These lignins were successfully used as coagulating agents for dyes, removing up to 90% of ethyl violet from simulated solutions (Gao et al. 2019).
Sulfonated lignins produced under normal pressure and temperature conditions were 96.9% soluble in water at neutral pH. By adding 1.49 mmol g-1 of sulfur in the lignin structure, sulfonation increased its negative zeta potential and improved its adsorption capacity of cement particles, leading to a better dispersion effect and homogeneity of the cement matrix (Ouyang et al. 2009).
CURRENT MARKET
Although kraft pulping is the largest lignin feedstock source worldwide, lignin from sulfite pulping is currently more commercially important because lignosulfonates have realistic applications, while kraft lignin is practically all burned for heating and power generation. Nevertheless, promising applications are opening up for kraft lignins due to the significant volumes produced worldwide.

Fig. 12. Lignin commercial brands and their applications by the prominent companies
Companies such as Ingevity, West Fraser, Domtar, RISE LignoDemo AB, WestRock, Suzano, and Liquid Lignin are dedicated to kraft lignin valorization. Likewise, Borregaard, Sappi Biotech GmbH, Domsjo Fabriker AB, Nippon Papers, and Rayonier Advanced Materials invest in technologies to produce lignosulfonates (Fig. 12). Together, these companies upgrade 1.8 million tons of lignin and lignin-derivatives per year, almost all at the commercial scale. Lignosulfonates supply ~79% of the lignin market (except for combustion), with minor contributions of kraft (~16%) and organosolv and hydrolysis (~5%) lignins (Dessbesell et al. 2020).
Borregaard (Sarpsborg, Norway), the major lignosulfonate producer worldwide, is present in four continents and delivers sustainable lignin-based materials, such as binding agents for animal feed, briquetting, dispersing agents for concrete, textile dyes, pesticides, battery components, and ceramic materials (Borregaard 2021).
After Borregaard, Domsjö Fabriker (Aditya Birla Group, Örnsköldsvik, Sweden), a prominent biorefinery facility, produces around 120,000 tons per year of powder lignin. Lignin is primarily sold for the external market to be used as an additive in concrete and feed industries, agriculture, and as a stabilizer for unpaved roads. Aditya Birla Group is a multinational business comprised of 50 companies with 130 production units in almost 40 countries (Domsjö 2021).
Sappi Biotech GmbH (Johannesburg, South Africa) developed technologies to apply lignosulfonates in leather tanning, dust control, recyclable packaging, water treatment as dispersing agents, and synthesis of phenolic resins. Besides, their lignosulfonates can be used as polyols and additives for ceramics, refractory materials, clay bricks, and bonding agents in fertilizers for soil improvements (SAPPI 2021).
Nippon Papers (Tokyo, Japan) takes advantage of lignin as dispersing, binding and chelating agents, selling around 70,000 tons per year of industrial brands such as SAN X®, VANILLEX®, and PEARLLEX®. These are lignosulfonate-based products that interact with various polymeric matrixes due to their abundance of functional groups and structural versatility (Nippon Papers 2021).
Rayonier Advanced Materials (Shelton, USA) produces ARBOTM for use as binding, chelating, and dispersants. Its lignin-based production is close to 130,000 tons per year in facilities located in the USA, France, and Canada (Rayonier, 2021). Also, both the Burgo Group (Verzuolo, Italy) and Shenyang Xingzhenghe Chemical (Shenyang, China) make the material available in the form of civil construction components. The latter company markets lignosulfonates in chrome, ferrochrome, potassium, calcium, and sodium forms. These materials have good properties for water reduction agents, adhesive, viscosity controllers, deflocculant, and fluid loss control agents (Shenyang Xingzhenghe Chemical 2021).
Concerning kraft lignin, Stora Enso (Helsinki, Finland), the oldest pulping company globally, started in 1288 as a copper mining company and diversified later to the forestry business. Today, it is the largest producer of commercial kraft lignin, with a production of circa 50,000 tons per year. Isolated by the LignoBoost process, this lignin branded as LineoTM is available with different dryness levels according to customer requirements for specific needs (Stora Enso 2021).
Ingevity (North Charleston, SC, USA), with an annual production of 40,000 tons per year, applies pine kraft lignin (Indulin AT) as the organic component in batteries. They also produce Kraftplex®, a kraft sodium lignosulfonate formulated to ensure better negative plate expanders for batteries. Besides, Ingevity’s kraft lignosulfonates are used to manufacture dispersants and surfactants to be applied in asphalt paving, agrochemicals, adhesives, lubricants, inks, coatings, elastomers, and bioplastics (Ingevity 2021).
Companies such as WestRock (formerly MeadWestvaco, Atlanta, GE, USA), Domtar (Fort Mill, USA), and Suzano (São Paulo, Brazil) operate with lignin production capacities of 30,000, 25,000, and 20,000 tons per year, respectively. The former has supplied kraft lignin to the market since the middle of the twentieth century. In 2014 Domtar started the commercial production of Biochoice™, a high-quality lignin based on the LignoBoost process. In this case, applications range from natural binders to fuels, additives, coatings, plastics, resins, and other carbon-based materials (Domtar 2021; WestRock 2021).
Since 2013, Suzano, a Brazilian pulp and paper mill in line with lignin supply companies, produces Ecolig, a renewable technologic platform of lignin-based products (Ligseal, Ligflow, Ligform, Ligflex, Ligsperse). Such products are applied as antioxidants in rubber compounds, dispersing agents and additives for concrete, and building blocks for phenolic resins and plastics (Suzano 2021).
Lignin is only second to cellulose as the most available biopolymer on Earth, and yet it is still primarily underutilized, despite the wide variety of products described above. Indeed, lignin valorization is in its infancy; however, following the example of mature companies, lignin can be the most valuable resource in biorefineries to produce sustainable, innovative, and high-performance biofuels biobased chemicals, and biomaterials.
FUTURE OUTLOOK AND PERSPECTIVES
A saying is widespread among people from wood pulping backgrounds, that “One can make everything from lignin except money” (Ragauskas et al. 2014). This thought is based on the fact that scientists can do almost everything starting from lignin, but no company has yet been able to transform these technologies into profitable products on a large scale.
Although no market for profitable businesses using technical lignin yet exists, the future perspectives are outstanding in the short term. Wood pulping companies are investing in isolation processes such as LignoBoost® and LignoForceTM to increase the availability of good quality kraft lignin for the market. New and exciting applications have been developed in recent years based on the research of numerous scientists.
The first point to be considered is which of the available technical lignin products is being referred to when discussing possible industrial applications. Pulping companies produce around 100 million metric tons per year of kraft and other technical lignins worldwide. However, less than 2% of these lignins are commercialized, mainly in adhesives, surfactants, and dispersant formulations (Bajwa et al. 2019). Lignosulfonates are by far the most important lignin derivative, with a production of 1.3 million metric tons per year. Regarding kraft lignin, just 265,000 metric tons are isolated and used today for biorefining applications. Also, soda lignin plus hydrolysis lignin from cellulosic ethanol production, with 75,000 metric tons per year, are increasing in their importance and market share (Dessbesell et al. 2020). Hence, these lignin sources must be studied in detail to give options for the development of new profitable businesses. Moreover, with investment much more remarkable than many billions of dollars, kraft lignin is by far the most important to be considered for emergent economies and developed countries (Li and Takkellapati 2018).
Lignin is crucial for developing a biorefinery concept based on circular bioeconomy and attending to the basic definitions of green chemistry and chemical engineering. Albeit sustainability is the primary concern, the lack of economic feasibility is the bottleneck, which is aggravated by its heterogeneity and wide molecular mass distribution. Fractionation may overcome part of this technical barrier, as well as functionatization to make lignin more uniform and suitable for a wide range of possible applications.
Among future products made from lignin, carbon fiber, bio-oil, and phenolic resins are leading applications, but binders, adhesives, bioplastics, concrete admixtures, and biomedical applications are on the rise (Bajwa et al. 2019). In the future, the cost-effectiveness of high-volume bulk applications (energy, phenolic resins, PUs) will benefit from the development of high value-added products such as antitumor drugs and other specialties such as nanomaterials, antioxidants, and renewable hydrocarbons (BTX and advanced biofuels), among others.
PUs and phenolic resins are the most critical applications for technical lignins in the near future. However, in the long term, judging by the research and development efforts expended to date, carbon fibers, and depolymerization processes to apply lignin as a source for aromatic chemicals will play an important role (Li and Takkellapati 2018).
Even for PU and phenolic resin applications, barriers still exist to commercialize the production technologies (Lettner et al. 2020). For PU resins, these include the quality assurance to support large-scale operations, potentially lower production costs and sales prices, supply security, and proven environmental benefits. For phenolic resins, the list includes competitive costs compared to the current market, reliability of the production process, supply security, proven environmental benefits, and the need for integration along the entire value chain. The least impacting parameters are lignin price, environmental benefits, supply security, and the high technology readiness level of process operations from this latter list. Therefore, the application of lignin in phenolic resins should become a reality very shortly.
The price of lignin residues from acid and enzymatic hydrolysis will be driven by the market for electricity, with minimum values being estimated around US$ 18 to 37 and US$ 43 to 70 per lignin ton, respectively (Obydenkova et al. 2019). In the case of phenolic resins, one may envisage that the cost of fossil phenol should limit the maximum price for lignin. On the other hand, for some special lignin applications in which high purity and batch-to-batch reliability are required (e.g., carbon fiber manufacturing), price might be less important than quality and compositional repeatability.
ACKNOWLEDGMENTS
The authors are grateful to CNPq (grant 309506/2017-4) for providing financial support to carry out this study. This work was also financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001.
REFERENCES CITED
Abu-Dalo, M. A., Al-Rawashdeh, N. A. F., and Ababneh, A. (2013). “Evaluating the performance of sulfonated Kraft lignin agent as corrosion inhibitor for iron-based materials in water distribution systems,” Desalination 313, 105-114. DOI: 10.1016/j.desal.2012.12.007
Alonso, M. V., Oliet, M., Rodríguez, F., García, J., Gilarranz, M. A., and Rodríguez, J. J. (2005). “Modification of ammonium lignosulfonate by phenolation for use in phenolic resins,” Bioresource Technology 96(9), 1013-1018. DOI: 10.1016/j.biortech.2004.09.009
Alwadani, N. S., and Fatehi, P. (2019). “Modification of kraft lignin with dodecyl glycidyl ether.” ChemistryOpen 8(10), 1258-1266. DOI: 10.1002/open.201900263
An, L., Si, C., Bae, J. H., Jeong, H., and Kim, Y. S. (2020). “One-step silanization and amination of lignin and its adsorption of Congo red and Cu(II) ions in aqueous solution,” International Journal of Biological Macromolecules 159, 222-230. DOI: 10.1016/j.ijbiomac.2020.05.072
Aro, T., and Fatehi, P. (2017). “Production and application of lignosulfonates and sulfonated lignin,” ChemSusChem 10(9), 1861-1877. DOI: 10.1002/cssc.201700082
Bajwa, D. S., Pourhashem, G., Ullah, A. H., and Bajwa, S. G. (2019). “A concise review of current lignin production, applications, products and their environment impact,” Industrial Crops and Products 139, 111526. DOI: 10.1016/j.indcrop.2019.111526
Berrima, B., Mortha, G., Boufi, S., Aloui, E. El, and Naceur Belgacem, M. (2016). “Oxypropylation of soda lignin: Characterization and application in polyurethane foams production,” Cellulose Chem. Technol. 50(9), 941-950.
Boerjan, W., Ralph, J., and Baucher, M. (2003). “Lignin biosynthesis,” Annual Review of Plant Biology 54(1), 519-546. DOI: 10.1146/annurev.arplant.54.031902.134938
Bolto, B., and Gregory, J. (2007). “Organic polyelectrolytes in water treatment,” Water Research 41(11), 2301-2324. DOI: 10.1016/j.watres.2007.03.012
Borregaard LignoTech, 2021. Industrial Applications. https://lignotech.com/Industrial-Applications (accessed 02.02.2021).
Brauns, F. E. (1939). “Native lignin I. Its isolation and methylation,” Journal of the American Chemical Society 61(8), 2120-2127. DOI: 10.1021/ja01877a043
Buono, P., Duval, A., Verge, P., Averous, L., and Habibi, Y. (2016). “New insights on the chemical modification of lignin: Acetylation versus silylation,” ACS Sustainable Chemistry and Engineering 4(10), 5212-5222. DOI: 10.1021/acssuschemeng.6b00903
Burgo Group, 2021. https://www.burgo.com/ (accessed 02.02.2021).
Bykov, G. L., and Ershov, B. G. (2010). “A sorbent based on phosphorylated lignin,” Russian Journal of Applied Chemistry 83(2), 316-319. DOI: 10.1134/S1070427210020254
Cateto, C. A., Barreiro, M. F., Rodrigues, A. E., and Belgacem, M. N. (2009). “Optimization study of lignin oxypropylation in view of the preparation of polyurethane rigid foams,” Industrial and Engineering Chemistry Research 48(5), 2583-2589. DOI: 10.1021/ie801251r
Chakar, F. S., and Ragauskas, A. J. (2004). “Review of current and future softwood kraft lignin process chemistry,” Industrial Crops and Products 20(2), 131-141. DOI: 10.1016/j.indcrop.2004.04.016
Chen, C., Zhu, M., Li, M., Fan, Y., and Sun, R. C. (2016). “Epoxidation and etherification of alkaline lignin to prepare water‑soluble derivatives and its performance in improvement of enzymatic hydrolysis efficiency,” Biotechnology for Biofuels 9(1), article no. 87. DOI: 10.1186/s13068-016-0499-9
Chen, X., Xi, X., Pizzi, A., Fredon, E., Du, G., Gerardin, C., and Amirou, S. (2020a). “Oxidized demethylated lignin as a bio-based adhesive for wood bonding,” Journal of Adhesion 97(9), 873-890. DOI: 10.1080/00218464.2019.1710830
Chen, Y. C., Fu, S., and Zhang, H. (2020b). “Signally improvement of polyurethane adhesive with hydroxy-enriched lignin from bagasse,” Colloids and Surfaces A: Physicochemical and Engineering Aspects 585, article no. 124164. DOI: 10.1016/j.colsurfa.2019.124164
Chen, Z., and Wan, C. (2018). “Ultrafast fractionation of lignocellulosic biomass by microwave-assisted deep eutectic solvent pretreatment,” Bioresource Technology 250, 532-537. DOI: 10.1016/j.biortech.2017.11.066
Cortés-Triviño, E., Valencia, C., Delgado, M. A., and Franco, J. M. (2018). “Modification of alkali lignin with poly(ethylene glycol) diglycidyl ether to be used as a thickener in bio-lubricant formulations,” Polymers 10(6), 670. DOI: 10.3390/polym10060670
Costes, L., Laoutid, F., Aguedo, M., Richel, A., Brohez, S., Delvosalle, C., and Dubois, P. (2016). “Phosphorus and nitrogen derivatization as efficient route for improvement of lignin flame retardant action in PLA,” European Polymer Journal 84, 652-667. DOI: 10.1016/j.eurpolymj.2016.10.003
Crestini, C., and Argyropoulos, D. S. (2002). “Structural analysis of wheat straw lignin by quantitative 31P and 2D NMR spectroscopy. The occurrence of ester bonds and α-O-4 substructures,” Journal of Agricultural and Food Chemistry 45(4), 1212-1219. DOI: 10.1021/jf960568k
Demuner, I. F., Colodette, J. L., Demuner, A. J., and Jardim, C. M. (2019). “Biorefinery review: Wide-reaching products through kraft lignin,” BioResources 14(3), 7543-7581. DOI: 10.15376/biores.14.3.Demuner
Dessbesell, L., Paleologou, M., Leitch, M., Pulkki, R., and Xu, C. (2020). “Global lignin supply overview and kraft lignin potential as an alternative for petroleum-based polymers,” Renewable and Sustainable Energy Reviews 123, article no. 109768. DOI: 10.1016/j.rser.2020.109768
Dilling, P. (1991). “Sulfonation of lignins,” Patent Number: 5,049,661, USA.
Domsjö (2021). Products. http://www.domsjo.adityabirla.com/en/Sidor/Lignin.aspx (accessed 02.02.2021).
Domtar (2021). “A biobased alternative to petroleum products,” https://www.domtar.com/en/what-we-make/biomaterials/biochoice-lignin (accessed 02.02.2021).
Du, X., Li, J., and Lindström, M. E. (2014). “Modification of industrial softwood kraft lignin using Mannich reaction with and without phenolation pretreatment,” Industrial Crops and Products 52, 729-735. DOI: 10.1016/j.indcrop.2013.11.035
Duval, A., and Avérous, L. (2020). “Mild and controlled lignin methylation with trimethyl phosphate: Towards a precise control of lignin functionality,” Green Chemistry 22(5), 1671-1680. DOI: 10.1039/C9GC03890F
Eraghi Kazzaz, A., Hosseinpour Feizi, Z., and Fatehi, P. (2019). “Grafting strategies for hydroxy groups of lignin for producing materials,” Green Chemistry 21(21), 5714-5752. DOI: 10.1039/C9GC02598G
Filley, T. R., Cody, G. D., Goodell, B., Jellison, J., Noser, C., and Ostrofsky, A. (2002). “Lignin demethylation and polysaccharide decomposition in spruce sapwood degraded by brown rot fungi,” Organic Geochemistry 33(2), 111-124. DOI: 10.1016/S0146-6380(01)00144-9
Francis, R. C., Bolton, T. S., Abdoulmoumine, N., Lavrykova, N., and Bose, S. K. (2008). “Positive and negative aspects of soda/anthraquinone pulping of hardwoods,” Bioresource Technology 99(17), 8453-8457. DOI: 10.1016/j.biortech.2008.02.055
Gao, C., Zhou, L., Yao, S., Qin, C., and Fatehi, P. (2020). “Phosphorylated kraft lignin with improved thermal stability,” International Journal of Biological Macromolecules 162, 1642-1652. DOI: 10.1016/j.ijbiomac.2020.08.088
Gao, W., Inwood, J. P. W., and Fatehi, P. (2019). “Sulfonation of phenolated kraft lignin to produce water soluble products,” Journal of Wood Chemistry and Technology 39(4), 225-241. DOI: 10.1080/02773813.2019.1565866
Ge, Y., Song, Q., and Li, Z. (2015). “A Mannich base biosorbent derived from alkaline lignin for lead removal from aqueous solution,” Journal of Industrial and Engineering Chemistry 23, 228-234. DOI: 10.1016/j.jiec.2014.08.021
Gouveia, J. R., Garcia, G. E. S., Antonino, L. D., Tavares, L. B., and Dos Santos, D. J. (2020). “Epoxidation of kraft lignin as a tool for improving the mechanical properties of epoxy adhesive,” Molecules 25(11), 1-18. DOI: 10.3390/molecules25112513
Graglia, M., Pampel, J., Hantke, T., Fellinger, T. P., and Esposito, D. (2016). “Nitro lignin-derived nitrogen-doped carbon as an efficient and sustainable electrocatalyst for oxygen reduction,” ACS Nano 10(4), 4364-4371. DOI: 10.1021/acsnano.5b08040
Hart, P. W., and Rudie, A. W. (2014). “Anthraquinone – A review of the rise and fall of a pulping catalyst,” Tappi Journal 13(10), 23-31. DOI: 10.32964/TJ13.10.23
He, W., and Fatehi, P. (2015). “Preparation of sulfomethylated softwood kraft lignin as a dispersant for cement admixture,” RSC Advances 5(58), 47031-47039. DOI: 10.1039/C5RA04526F
He, X., Luzi, F., Yang, W., Xiao, Z., Torre, L., Xie, Y., and Puglia, D. (2018). “Citric acid as green modifier for tuned hydrophilicity of surface modified cellulose and lignin nanoparticles,” ACS Sustainable Chemistry and Engineering 6(8), 9966-9978. DOI: 10.1021/acssuschemeng.8b01202
Hopa, D. Y., and Fatehi, P. (2020). “Using sulfobutylated and sulfomethylated lignin as dispersant for kaolin suspension,” Polymers 12(9), 11-14. DOI: 10.3390/polym12092046
Hu, J., Zhang, Q., and Lee, D. J. (2018). “Kraft lignin biorefinery: A perspective,” Bioresource Technology 247, 1181-1183. DOI: 10.1016/j.biortech.2017.08.169
Hu, L., Pan, H., Zhou, Y., Hse, C. Y., Liu, C., Zhang, B., and Xu, B. (2014). “Chemical groups and structural characterization of lignin via thiol-mediated demethylation,” Journal of Wood Chemistry and Technology 34(2), 122-134. DOI: 10.1080/02773813.2013.844165
Hu, L., Pan, H., Zhou, Y., and Zhang, M. (2011). “Methods to improve lignin’s reactivity as a phenol substitute and as replacement for other phenolic compounds: A brief review,” BioResources, 6(3), 3515-3525. DOI: 10.15376/biores.6.3.3515-3525
Huang, C., Ma, J., Zhang, W., Huang, G., and Yong, Q. (2018). “Preparation of lignosulfonates from biorefinery lignins by sulfomethylation and their application as a water reducer for concrete,” Polymers 10(8), article no. 481. DOI: 10.3390/polym10080841
Ingevity (2021). Products. https://www.ingevity.com/products/kraftplex/ (accessed 02.02.2021).
Jablonskis, A., Arshanitsa, A., Arnautov, A., Telysheva, G., and Evtuguin, D. (2018). “Evaluation of Ligno BoostTM softwood kraft lignin epoxidation as an approach for its application in cured epoxy resins,” Industrial Crops and Products 112, 225-235. DOI: 10.1016/j.indcrop.2017.12.003
Jiang, X., Liu, J., Du, X., Hu, Z., Chang, H. M., and Jameel, H. (2018). “Phenolation to improve lignin reactivity toward thermosets application,” ACS Sustainable Chemistry and Engineering 6(4), 5504-5512. DOI: 10.1021/acssuschemeng.8b00369
Jiang, Y., Feng, Y., Lei, B., and Zhong, H. (2020). “Impact mechanisms of supercritical CO2–ethanol–water on extraction behavior and chemical structure of eucalyptus lignin,” International Journal of Biological Macromolecules 161, 1506-1515. DOI: 10.1016/j.ijbiomac.2020.07.318
Kamimura, N., Sakamoto, S., Mitsuda, N., Masai, E., and Kajita, S. (2019). “Advances in microbial lignin degradation and its applications,” Current Opinion in Biotechnology 56, 179-186. DOI: 10.1016/j.copbio.2018.11.011
Kamimura, N., Takahashi, K., Mori, K., Araki, T., Fujita, M., Higuchi, Y., and Masai, E. (2017). “Bacterial catabolism of lignin-derived aromatics: New findings in a recent decade: Update on bacterial lignin catabolism,” Environmental Microbiology Reports 9(6), 679-705. DOI: 10.1111/1758-2229.12597
Kamoun, A., Jelidi, A., and Chaabouni, M. (2003). “Evaluation of the performance of sulfonated esparto grass lignin as a plasticizer-water reducer for cement,” Cement and Concrete Research 33, 995-1003. DOI: 10.1016/S0008-8846(02)01098-0
Kim, J. Y., Hafezi-Sefat, P., Cady, S., Smith, R. G., and Brown, R. C. (2019). “Premethylation of lignin hydroxyl functionality for improving storage stability of oil from solvent liquefaction,” Energy and Fuels 33(2), 1248-1255. DOI: 10.1021/acs.energyfuels.8b03894
Klein, S. E., Alzagameem, A., Rumpf, J., Korte, I., Kreyenschmidt, J., and Schulze, M. (2019). “Antimicrobial activity of lignin-derived polyurethane coatings prepared from unmodified and demethylated lignins,” Coatings 9(8), article no. 494. DOI: 10.3390/coatings9080494
Ko, H. U., Kim, J. W., Kim, H. C., Zhai, L., and Kim, J. (2020). “Esterified PVA-lignin resin by maleic acid applicable for natural fiber reinforced composites,” Journal of Applied Polymer Science 137(26), article no. 48836. DOI: 10.1002/app.48836
Konduri, M. K. R., and Fatehi, P. (2015). “Production of water-soluble hardwood kraft lignin via sulfomethylation using formaldehyde and sodium sulfite,” ACS Sustainable Chemistry and Engineering 3(6), 1172-1182. DOI: 10.1021/acssuschemeng.5b00098
Kouisni, L., Gagné, A., Maki, K., Holt-Hindle, P., and Paleologou, M. (2016). “LignoForce system for the recovery of lignin from black liquor: Feedstock options, odor profile, and product characterization,” ACS Sustainable Chemistry and Engineering 4(10), 5152-5159. DOI: 10.1021/acssuschemeng.6b00907
Kühnel, I., Saake, B., and Lehnen, R. (2017). “Comparison of different cyclic organic carbonates in the oxyalkylation of various types of lignin,” Reactive and Functional Polymers 120, 83-91. DOI: 10.1016/j.reactfunctpolym.2017.09.011
Lakhmanov, D. E., Khabarov, Y. G., Veshnyakov, V. A., and Yokubjanov, M. R. (2020). “Nitration of hydrolysis lignin in water-aprotic solvent mixtures,” Forestry Journal 377(5), 184-192. DOI: 10.37482/0536-1036-2020-5-184-192
Lee, Y., Park, C. H., and Lee, E. Y. (2017). “Chemical modification of methanol-insoluble kraft lignin using oxypropylation under mild conditions for the preparation of bio-polyester,” Journal of Wood Chemistry and Technology 37(5), 334-342. DOI: 10.1080/02773813.2017.1303512
Lettner, M., Hesser, F., Hedeler, B., Schwarzbauer, P., and Stern, T. (2020). “Barriers and incentives for the use of lignin-based resins: Results of a comparative importance performance analysis,” Journal of Cleaner Production 256, article no. 120520. DOI: 10.1016/j.jclepro.2020.120520
Li, J., Wang, W., Zhang, S., Gao, Q., Zhang, W., and Li, J. (2016). “Preparation and characterization of lignin demethylated at atmospheric pressure and its application in fast curing biobased phenolic resins Jiongjiong,” RSC Advances 6, 67435-67443. DOI: 10.1039/C6RA11966B
Li, J., Zhang, J., Zhang, S., Gao, Q., Li, J., and Zhang, W. (2017). “Fast curing bio-based phenolic resins via lignin demethylated under mild reaction condition,” Polymers 9(9), article no. 428. DOI: 10.3390/polym9090428
Li, T., and Takkellapati, S. (2018). “The current and emerging sources of technical lignins and their applications,” Biofuels, Bioproducts and Biorefining 12(5), 756-787. DOI: 10.1002/bbb.1913
Li, T., Yin, Y., Wu, S., Ma, H., and Zhang, F. (2020). “Effect of pre-acetylation of hydroxyl functional groups by choline chloride/acetic anhydride on subsequent lignin pyrolysis,” Bioresource Technology 317, article no. 124034. DOI: 10.1016/j.biortech.2020.124034
Li, Y., and Ragauskas, A. J. (2012). “Ethanol organosolv lignin-based rigid polyurethane foam reinforced with cellulose nanowhiskers,” RSC Advances 2(8), 3347-3351. DOI: 10.1039/c2ra00646d
Li, Z., and Ge, Y. (2011). “Extraction of lignin from sugar cane bagasse and its modification into a high performance dispersant for pesticide formulations,” Journal of the Brazilian Chemical Society 22(10), 1866-1871. DOI: 10.1590/S0103-50532011001000006
Liu, L. Y., Hua, Q., and Renneckar, S. (2019). “A simple route to synthesize esterified lignin derivatives,” Green Chemistry 21(13), 3682-3692. DOI: 10.1039/C9GC00844F
Liu, X., Zhu, H., Qin, C., Zhou, J., Zhao, J. R., and Wang, S. (2013). “Adsorption of heavy metal ion from aqueous single metal solution by aminated epoxy-lignin,” BioResources 8(2), 2257-2269. DOI: 10.15376/biores.8.2.2257-2269
Liu, Z., Lu, X., An, L., and Xu, C. (2016). “A novel cationic lignin-amine emulsifier with high performance reinforced via phenolation and mannich reactions,” BioResources 11(3), 6438-6451. DOI: 10.15376/biores.11.3.6438-6451
Londoño Zuluaga, C., Du, J., Chang, H.-M., Jameel, H., and Gonzalez, R. W. (2018). “Lignin modifications and perspectives towards applications of phenolic foams: A review,” BioResources 13(4), 9158-9179. DOI: 10.15376/biores.13.4.Londono-Zuluaga
Lou, H., Lai, H., Wang, M., Pang, Y., Yang, D., Qiu, X., Wang, B., and Zhang, H. (2013). “Preparation of lignin-based superplasticizer by graft sulfonation and investigation of the dispersive performance and mechanism in a cementitious system,” Industrial and Engineering Chemistry Research 52(46), 16101-16109. DOI: 10.1021/ie402169g
Luo, S., Cao, J., and McDonald, A. G. (2018). “Cross-linking of technical lignin via esterification and thermally initiated free radical reaction,” Industrial Crops and Products 121(5), 169-179. DOI: 10.1016/j.indcrop.2018.05.007
Ma, H., Li, T., Wu, S., and Zhang, X. (2021). “Demethylation of a methoxy group to inhibit repolymerization during alkaline lignin pyrolysis,” Fuel 286, article no. 119394. DOI: 10.1016/j.fuel.2020.119394
Marqueño, A., Pérez-Albaladejo, E., Flores, C., Moyano, E., and Porte, C. (2019). “Toxic effects of bisphenol A diglycidyl ether and derivatives in human placental cells,” Environmental Pollution 244, 513-521. DOI: 10.1016/j.envpol.2018.10.045
Matsushita, Y., Hirano, D., Aoki, D., Yagami, S., Takagi, Y., and Fukushima, K. (2017). “A biobased flame-retardant resin based on lignin,” Advanced Sustainable Systems 1(10), article no. 1700073. DOI: 10.1002/adsu.201700073
Meister, J. J. (2002). “Modification of lignin,” Journal of Macromolecular Science, Part C: Polymer Reviews 42(2), 235-289. DOI: 10.1081/MC-120004764
Miki, K., Kamitakahara, H., Yoshinaga, A., Tobimatsu, Y., and Takano, T. (2020). “Methylation-triggered fractionation of lignocellulosic biomass to afford cellulose-, hemicellulose-, and lignin-based functional polymers: Via click chemistry,” Green Chemistry 22(9), 2909-2928. DOI: 10.1039/D0GC00451K
Mou, H., Huang, J., Li, W., Wu, X., Liu, Y., and Fan, H. (2020). “Study on the chemical modification of alkali lignin towards for cellulase adsorbent application,” International Journal of Biological Macromolecules 149, 794-800. DOI: 10.1016/j.ijbiomac.2020.01.229
Mutturi, S., Palmqvist, B., and Lidén, G. (2014). “Developments in bioethanol fuel-focused biorefineries,” in: Advances in Biorefineries: Biomass and Waste Supply Chain Exploitation, K. Waldron (ed.), Woodhead Publishing, Sweden, pp. 259-302. DOI: 10.1533/9780857097385.1.259
Nada, A. A. M. A., and Hassan, M. L. (2003). “Phosphorylated cation-exchangers from cotton stalks and their constituents,” Journal of Applied Polymer Science 89(11), 2950-2956. DOI: 10.1002/app.12408
Nasiri, A., Wearing, J., and Dubé, M. A. (2020). “The use of lignin in emulsion-based pressure-sensitive adhesives,” International Journal of Adhesion and Adhesives 100, article no. 102598. DOI: 10.1016/j.ijadhadh.2020.102598
Nikafshar, S., Zabihi, O., Moradi, Y., Ahmadi, M., Amiri, S., and Naebe, M. (2017). “Catalyzed synthesis and characterization of a novel lignin-based curing agent for the curing of high-performance epoxy resin,” Polymers 9(7), article no. 266. DOI: 10.3390/polym9070266
Nippon Papers, 2021. Lignin Products. https://www.nipponpapergroup.com/english/products/chemical/lignin_products/index.html (accessed 02.02.2021).
Obydenkova, S. V., Kouris, P. D., Hensen, E. J. M., Smeulders, D. M. J., van der Meer, Y., and Boot, M. D. (2019). “Industrial lignin from 2G biorefineries – Assessment of availability and pricing strategies,” Bioresource Technology 291, article no. 121805. DOI: 10.1016/j.biortech.2019.121805
de Oliveira, D. R., Avelino, F., Mazzetto, S. E., and Lomonaco, D. (2020). “Microwave-assisted selective acetylation of Kraft lignin: Acetic acid as a sustainable reactant for lignin valorization,” International Journal of Biological Macromolecules 164, 1536-1544. DOI: 10.1016/j.ijbiomac.2020.07.216
Ouyang, X., Ke, L., Qiu, X., Guo, Y., and Pang, Y. (2009). “Sulfonation of alkali lignin and its potential use in dispersant for cement,” Journal of Dispersion Science and Technology 30(1), 1-6. DOI: 10.1080/01932690802473560
Pan, H., Sun, G., and Zhao, T. (2013). “Synthesis and characterization of aminated lignin,” International Journal of Biological Macromolecules 59, 221-226. DOI: 10.1016/j.ijbiomac.2013.04.049
Pang, Y., Chen, Z., Zhao, R., Yi, C., Qiu, X., Qian, Y., and Lou, H. (2021). “Facile synthesis of easily separated and reusable silver nanoparticles/aminated alkaline lignin composite and its catalytic ability,” Journal of Colloid And Interface Science 587, 334-346. DOI: 10.1016/j.jcis.2020.11.113
Podschun, J., Saake, B., and Lehnen, R. (2017). “Catalytic demethylation of organosolv lignin in aqueous medium using indium triflate under microwave irradiation,” Reactive and Functional Polymers 119, 82-86. DOI: 10.1016/j.reactfunctpolym.2017.08.007
Prieur, B., Meub, M., Wittemann, M., Klein, R., Bellayer, S., Fontaine, G., and Bourbigot, S. (2016). “Phosphorylation of lignin to flame retard acrylonitrile butadiene styrene (ABS),” Polymer Degradation and Stability 127, 32-43. DOI: 10.1016/j.polymdegradstab.2016.01.015
Qin, Y., Yang, D., Guo, W., and Qiu, X. (2015). “Investigation of grafted sulfonated alkali lignin polymer as dispersant in coal-water slurry,” Journal of Industrial and Engineering Chemistry, The Korean Society of Industrial and Engineering Chemistry, 27, 192-200. DOI: 10.1016/j.jiec.2014.12.034
Ragauskas, A. J., Beckham, G. T., Biddy, M. J., Chandra, R., Chen, F., Davis, M. F., Davison, B. H., Dixon, R. A., Gilna, P., Keller, M., Langan, P., Naskar, A. K., Saddler, J. N., Tschaplinski, T. J., Tuskan, G. A., and Wyman, C. E. (2014). “Lignin valorization: Improving lignin processing in the biorefinery,” Science 344, article no. 1246843. DOI: 10.1126/science.1246843
Ramos, L. P., Suota, M. J., Fockink, D. H., Pavaneli, G., da Silva, T. A., and Łukasik, R. M. (2020). “Enzymes and biomass pretreatment,” in: Recent Advances in Bioconversion of Lignocellulose to Biofuels and Value-Added Chemicals within the Biorefinery Concept, E. X. Ferreira Filho, E. A. Ximenes, L. R. S. Moreira, C. S. Farinas (eds.), Elsevier, pp. 61-100. DOI: 10.1016/C2018-0-03277-6
Rashid, T., Sher, F., Rasheed, T., Zafar, F., Zhang, S., and Murugesan, T. (2021). “Evaluation of current and future solvents for selective lignin dissolution – A review,” Journal of Molecular Liquids 321, article no. 114577. DOI: 10.1016/j.molliq.2020.114577
Rayonier (2021). Products. https://rayonieram.com/facilities/ (accessed 02.02.2021).
Sadeghifar, H., Cui, C., and Argyropoulos, D. S. (2012). “Toward thermoplastic lignin polymers. Part 1. Selective masking of phenolic hydroxyl groups in kraft lignins via methylation and oxypropylation chemistries,” Industrial and Engineering Chemistry Research 51(51), 16713-16720. DOI: 10.1021/ie301848j
Sameni, J., Krigstin, S., and Sain, M. (2017). “Solubility of lignin and acetylated lignin in organic solvents,” BioResources 12(1), 1548-1565. DOI: 10.15376/biores.12.1.1548-1565
SAPPI (2021). “Lignin application areas,” https://www.sappi.com/lignin-application-areas-0 (accessed 02.02.2021).
Saraf, V. P., and Glasser, W. G. (1984). “Engineering plastics from lignin. III. Structure property relationships in solution cast polyurethane films,” Journal of Applied Polymer Science 29(5), 1831-1841. DOI: 10.1002/app.1984.070290534
Sawamura, K., Tobimatsu, Y., Kamitakahara, H., and Takano, T. (2017). “Lignin functionalization through chemical demethylation: preparation and tannin-like properties of demethylated guaiacyl-type synthetic lignins,” ACS Sustainable Chemistry and Engineering 5(6), 5424-5431. DOI: 10.1021/acssuschemeng.7b00748
Schutyser, W., Renders, T., Van Den Bosch, S., Koelewinjn, S.-F., G. T. Beckham, and Sels, B. F. (2018). “Chemicals from lignin: An interplay of lignocellulose fractionation, depolymerisation, and upgrading,” Royal Society of Chemistry 47, 852. DOI: 10.1039/C7CS00566K
Sen, S., Patil, S., and Argyropoulos, D. S. (2015). “Methylation of softwood kraft lignin with dimethyl carbonate,” Green Chemistry 17(2), 1077-1087. DOI: 10.1039/C4GC01759E
Shayesteh, K., Mohammadzadeh, G., and Zamanloo, M. (2020). “Study and optimization of parameters affecting the acetylation process of lignin sulfonate biopolymer,” International Journal of Biological Macromolecules 163, 1810-1820. DOI: 10.1016/j.ijbiomac.2020.09.014
Shen, X., Meng, Q., Mei, Q., Xiang, J., Liu, H., and Han, B. (2020). “The production of 4-ethyltoluene: Via directional valorization of lignin,” Green Chemistry 22(7), 2191-2196. DOI: 10.1039/D0GC00587H
Shenyang Xingzhenghe Chemical (http://xzhch.com/products/lignosulfonate/ (accessed 02.02.2021).
Song, Y., Wang, Z., Yan, N., Zhang, R., and Li, J. (2016). “Demethylation of wheat straw alkali lignin for application in phenol formaldehyde adhesives,” Polymers 8(6), 1-14. DOI: 10.3390/polym8060209
Stora Enso (2021). Lineo by Stora Enso. https://www.storaenso.com/en/products/lignin/lineo (accessed 02.02.2021).
Suzano (2021). https://www.suzanoecolig.com.br/ (accessed 02.02.2021).
Taleb, F., Ammar, M., Mosbah, M. ben, Salem, R. ben, and Moussaoui, Y. (2020). “Chemical modification of lignin derived from spent coffee grounds for methylene blue adsorption,” Scientific Reports 10(1), 1-14. DOI: 10.1038/s41598-020-68047-6
Thébault, M., Kutuzova, L., Jury, S., Eicher, I., Zikulnig-Rusch, E. M., and Kandelbauer, A. (2020). “Effect of phenolation, lignin-type and degree of substitution on the properties of lignin-modified phenol-formaldehyde impregnation resins: Molecular weight distribution, wetting behavior, rheological properties and thermal curing profiles,” Journal of Renewable Materials 8(6), 603-630. DOI: 10.32604/jrm.2020.09616
Venkatesagowda, B. (2019). “Enzymatic demethylation of lignin for potential biobased polymer applications,” Fungal Biology Reviews 33(3–4), 190-224. DOI: 10.1016/j.fbr.2019.06.002
Venkatesagowda, B., and Dekker, R. F. H. (2021). “Enzyme and microbial technology microbial demethylation of lignin: Evidence of enzymes participating in the removal of methyl/methoxyl groups,” Enzyme and Microbial Technology 147, article no. 109780. DOI: 10.1016/j.enzmictec.2021.109780
Wang, B., Chen, T. Y., Wang, H. M., Li, H. Y., Liu, C. F., and Wen, J. L. (2018a). “Amination of biorefinery technical lignins using Mannich reaction synergy with subcritical ethanol depolymerization,” International Journal of Biological Macromolecules 107, 426-435. DOI: 10.1016/j.ijbiomac.2017.09.012
Wang, F., Kuai, J., Pan, H., Wang, N., and Zhu, X. (2018b). “Study on the demethylation of enzymatic hydrolysis lignin and the properties of lignin–epoxy resin blends,” Wood Science and Technology 52(5), 1343-1357. DOI: 10.1007/s00226-018-1024-z
Wang, L., Lagerquist, L., Zhang, Y., Koppolu, R., Tirri, T., Sulaeva, I., Schoultz, S. von, Vähäsalo, L., Pranovich, A., Rosenau, T., Eklund, P. C., Willför, S., Xu, C., and Wang, X. (2020). “Tailored thermosetting wood adhesive based on well-defined hardwood lignin fractions,” ACS Sustainable Chemistry and Engineering 8(35), 13517-13526. DOI: 10.1021/acssuschemeng.0c05408
Wang, X., Zhang, Y., Hao, C., Dai, X., Zhou, Z., and Si, N. (2014). “Ultrasonic-assisted synthesis of aminated lignin by a Mannich reaction and its decolorizing properties for anionic azo-dyes,” RSC Advances 4(53), 28156-28164. DOI: 10.1039/c4ra03133d
Wang, Y. Y., Chen, Y. R., and Sarkanen, S. (2015). “Path to plastics composed of ligninsulphonates (lignosulfonates).” Green Chemistry 17(11), 5069-5078. DOI: 10.1039/C5GC01865J
WestRock (2021). https://www.westrock.com/ (accessed 02.02.2021).
Wu, H., Chen, F., Feng, Q., and Yue, X. (2012). “Oxidation and sulfomethylation of alkali-extracted lignin from corn stalk,” BioResources 7(3), 2742-2751.
Wu, L. C.-F., and Glasser, W. G. (1984). “Engineering plastics from lignin. I. Synthesis of hydroxypropyl lignin,” Journal of Applied Polymer Science 29, 1111-1123. DOI: 10.1002/app.1984.070290408
Xiang, Q., and Xiao, F. (2020). “Applications of epoxy materials in pavement engineering,” Construction and Building Materials 235, article no. 117529. DOI: 10.1016/j.conbuildmat.2019.117529
Xiong, S. J., Pang, B., Zhou, S. J., Li, M. K., Yang, S., Wang, Y. Y., Shi, Q., Wang, S. F., Yuan, T. Q., and Sun, R. C. (2020). “Economically competitive biodegradable PBAT/lignin composites: Effect of lignin methylation and compatibilizer,” ACS Sustainable Chemistry and Engineering 8(13), 5338-5346. DOI: 10.1021/acssuschemeng.0c00789
Xu, J., Zhu, S., Liu, P., Gao, W., Li, J., and Mo, L. (2017). “Adsorption of Cu(II) ions in aqueous solution by aminated lignin from enzymatic hydrolysis residues,” RSC Advances 7(71), 44751-44758. DOI: 10.1039/C7RA06693G
Yang, L., Wang, X. F., Cui, Y., Tian, Y. M., Chen, H. Z., and Wang, Z. C. (2014). “Modification of renewable resources — lignin — by three chemical methods and its applications to polyurethane foams,” Polymers for Advanced Technologies 25(10), 1089-1098. DOI: 10.1002/pat.3356
Yetiş, F., Liu, X., Sampson, W. W., and Gong, R. H. (2020). “Acetylation of lignin containing microfibrillated cellulose and its reinforcing effect for polylactic acid,” European Polymer Journal 134, article no. 109803. DOI: 10.1016/j.eurpolymj.2020.109803
Yu, Y., Fu, S., Song, P., Luo, X., Jin, Y., Lu, F., Wu, Q., and Ye, J. (2012). “Functionalized lignin by grafting phosphorus-nitrogen improves the thermal stability and flame retardancy of polypropylene,” Polymer Degradation and Stability 97(4), 541-546. DOI: 10.1016/j.polymdegradstab.2012.01.020
Zhang, F., Jiang, X., Lin, J., Zhao, G., Chang, H. M., and Jameel, H. (2019a). “Reactivity improvement by phenolation of wheat straw lignin isolated from a biorefinery process,” New Journal of Chemistry 43(5), 2238-2246. DOI: 10.1039/C8NJ05016C
Zhang, J., Lin, X., Luo, X., Zhang, C., and Zhu, H. (2011). “A modified lignin adsorbent for the removal of 2,4,6-trinitrotoluene,” Chemical Engineering Journal 168(3), 1055-1063. DOI: 10.1016/j.cej.2011.01.083
Zhang, L., and Huang, J. (2001). “Effects of nitrolignin on mechanical properties of polyurethane-nitrolignin films,” Journal of Applied Polymer Science 80(8), 1213-1219. DOI: 10.1002/app.1206
Zhang, M., and Ogale, A. A. (2014). “Carbon fibers from dry-spinning of acetylated softwood kraft lignin,” Carbon 69, 626-629. DOI: 10.1016/j.carbon.2013.12.015
Zhang, X., Kim, Y., Elsayed, I., Taylor, M., Eberhardt, T. L., Hassan, E. B., and Shmulsky, R. (2019b). “Rigid polyurethane foams containing lignin oxyalkylated with ethylene carbonate and polyethylene glycol,” Industrial Crops and Products 141, article no. 111797. DOI: 10.1016/j.indcrop.2019.111797
Zhou, M., Shi, H., Li, C., Sheng, X., Sun, Y., Hou, M., Niu, M., and Pan, X. (2020). “Depolymerization and activation of alkali lignin by solid acid-catalyzed phenolation for preparation of lignin-based phenolic foams,” Industrial and Engineering Chemistry Research 59(32), 14296-14305. DOI: 10.1021/acs.iecr.0c01753
Zhu, G., Qiu, X., Zhao, Y., Qian, Y., Pang, Y., and Ouyang, X. (2016). “Depolymerization of lignin by microwave-assisted methylation of benzylic alcohols,” Bioresource Technology 218, 718-722. DOI: 10.1016/j.biortech.2016.07.021
Article submitted: April 15, 2021; Peer review completed: May 15, 2021; Revised version received: June 16, 2021; Accepted: July 4, 2021; Published: July 12, 2021.
DOI: 10.15376/biores.16.3.Suota