
Abstract
Pinus pinaster wood was pulped in ethanol/water medium catalyzed with sulfuric acid, and lignin was recovered from the liquid phase by precipitation upon water addition. Lignin samples were characterized for composition and thermal properties. Lignin samples were reacted with selected esterification agents (butyric, isobutyric, or crotonic anhydrides) under experimental conditions leading to extensive conversion of the available hydroxyl groups, and the esterified lignins were assessed for composition and thermal properties. Samples made up of crude polylactic acid or its blends with lignins (raw or esterified) were assayed for mechanical properties. The blends of polylactic acid with lignin modified with butyric anhydride presented higher Young’s modulus and elongation at break than neat polylactic acid.
Download PDF
Full Article
Manufacture, Characterization, and Properties of Poly-(lactic acid) and its Blends with Esterified Pine Lignin
Carlos Vila,a,b Valentín Santos,a,b Bodo Saake,c and Juan C. Parajó a,b,*
Pinus pinaster wood was pulped in ethanol/water medium catalyzed with sulfuric acid, and lignin was recovered from the liquid phase by precipitation upon water addition. Lignin samples were characterized for composition and thermal properties. Lignin samples were reacted with selected esterification agents (butyric, isobutyric, or crotonic anhydrides) under experimental conditions leading to extensive conversion of the available hydroxyl groups, and the esterified lignins were assessed for composition and thermal properties. Samples made up of crude polylactic acid or its blends with lignins (raw or esterified) were assayed for mechanical properties. The blends of polylactic acid with lignin modified with butyric anhydride presented higher Young’s modulus and elongation at break than neat polylactic acid.
Keywords: Lignin; Polylactic acid; Mechanical properties; Thermal properties
Contact information: a: Department of Chemical Engineering, Faculty of Science, University of Vigo (Campus Ourense), As Lagoas, 32004 Ourense, Spain: b: CITI (Centro de Investigación, Transferencia e Innovación) – University of Vigo, Tecnopole, San Cibrao das Viñas, Ourense, Spain; c: Department of Wood Science, Chemical Wood Technology, University of Hamburg, Leuschnerstraße 91b, 21031 Hamburg, Germany; *Corresponding author: jcparajo@uvigo.es
INTRODUCTION
The economic, environmental, and sustainability issues derived from the large-scale utilization of fossil resources as fuels or as raw materials for the industry fosters the transition to a bio-based economy, in which lignocellulosic materials are expected to play a key role.
The lignocellulose biorefinery concept, based on the chemical separation of the major components (cellulose, hemicellulose, and lignin) and their further individual processing, is an interesting approach to maximize the added value of the final products. Lignin valorization is one of the major problems involved in the development of biorefineries. Even if lignin is a large source of aromatic compounds, most of it is currently burned as a crude fuel, and only a small percentage is being used in other industrial applications. In order to get added value from lignin, high-volume applications based on its polymeric nature have been proposed, including the manufacture of sustainable plastics and composites (for example, as additives or fillers for polyolefin-based materials) (Pérez-Guerrero et al. 2014). Lignin has been employed in formulation of elastomeric and thermoset materials (Thielemans et al. 2002); but the use of lignin in the formulation of thermoplastic composites has been a challenge (Hilburg et al. 2014), due to their poor dispersion and compatibility (Toriz et al. 2002; Chung et al. 2013). To overcome these problems, the utilization of compatibilizers and/or chemically modified lignins has been proposed (Gordobil et al. 2014).
Since lignin has free aliphatic and phenolic hydroxyl groups, both esterification and etherification are potential reactions for chemical modification. The thermal and mechanical properties of blends of acetylated lignin with synthetic polymers have been assessed (Jeong et al. 2012; Gordobil et al. 2014). In general, both the compatibility of esterified lignin with plastics and the mechanical properties of the resulting composites depend on the specific materials employed.
Polylactic acid (PLA) is a biodegradable thermoplastic polymer with a growing market, which can be manufactured from renewable resources and processed in conventional equipment. PLA has a comparatively good susceptibility to thermal processing (Ouyang et al. 2012), and exhibits high strength and stiffness, but it has a brittle nature. Although specific PLA properties (including flexibility and ductility) can be enhanced by plasticization (Liu and Zhang 2011), some PLA-based composites have shown poor mechanical properties (Toriz et al. 2002; Li et al. 2003; Ouyang et al. 2012), whereas composites containing lignin and PLA have been reported to present increased thermal stability and impact strength (Spiridon et al. 2015). Gordobil et al. (2014) reported an increased thermal stability at a fairly constant tensile strength for composites containing acetylated lignins.
In this work, Pinus pinaster wood was subjected to ethanol-water pulping, and lignin was recovered by precipitation. The recovered lignin was characterized and subjected to esterification with three anhydrides of the same chain length. Lignin and modified lignins were blended with PLA, and the mechanical properties of the resulting materials were determined.
EXPERIMENTAL
Materials
Pinus pinaster wood chips were kindly provided by Orember-Finsa, Ourense (Spain). Samples were air-dried, then homogenized in a single lot to avoid compositional differences among samples and stored until use. The chemicals used for lignin esterification (1-methylimidazole, 1,4-dioxane, butyric anhydride, isobutyric anhydride, and crotonic anhydride) were purchased from Sigma-Aldrich. The corresponding structures are shown in Table 1.
Polylactic acid (Nature Works 3051 from Cargill Dow LLC, USA) presented the followings properties: density, 1.25 g/cm3; glass transition temperature (Tg), 59 ºC; melting temperature (Tm), 152 ºC; and melt flow index (MFI), 30.3 g/10 min (measured at 210 ºC and 21.2 N).
Organosolv Delignification
Pinus pinaster wood was processed at 175 ºC for 120 min in water/ethanol (1:1 v/v) using a liquid to solid ratio of 5, in the presence of 1% H2SO4. After cooking, the pulp was filtrated and washed with the same volume of hot water/ethanol (1:1) solution. The filtrates were combined, and water was added (2:1 v/v) to precipitate the organosolv lignin. The precipitated lignin was recovered by filtration, washed with hot water, and dried in an oven vacuum at 50 ºC up to constant weight.
Table 1. Esterification Agents and Solvents Used for Lignin Modification, and Results Obtained for the Mass Gain Percentage (MPG)

Lignin Esterification
Esterification reactions were performed according to the method of Thielemans and Wool (2005) modified as follows: lignin was suspended in the considered solvent at 2:1 weight ratio. In esterification reactions with butyric or isobutyric anhydrides, the anhydride itself was employed as a solvent, whereas the reactions with crotonic anhydride were performed in 1,4-dioxane (see Table 1). In this latter case, the mass ratio of lignin to crotonic anhydride was 1:1. N-methylimidazole (0.01 mL/g lignin) was employed as a catalyst. Reactions were performed in stirred glass reactors kept at 50 ºC for 3 h. After reaction, the solution was poured into cold deionized water, and the precipitated esterified lignin was recovered by filtration and washed with water until the filtrate reached pH = 5. Esterified lignins were oven-dried at 50 ºC under vacuum to constant weight, and employed to measure the mass gain percentage (MGP) respect the original lignin used in the reaction. The MGP data are listed in Table 1.
Analysis
Samples of Pinus pinaster wood, organosolv pulp, organosolv lignin (OL), and esterified lignins were subjected to quantitative acid hydrolysis with 72% (w/w) sulfuric acid (TAPPI-249-em-85 method). The solid residue after hydrolysis was recovered by filtration and considered as acid-insoluble lignin. The carbohydrate contents of hydrolyzates were measured by HPLC using an Agilent 1100 instrument fitted with a Refractive Index Detector and an Aminex HPX-87P column (BioRad, Life Science Group Hercules, CA). Acid-soluble lignin in hydrolyzates was determined spectrophotometrically at 205 nm (TAPPI UM250 um-83 method). The ash content was determined by incineration at 575 ºC for 8 to 12 h. Elemental analysis was performed using a Fisons Analyzer (Carlo Erba). NMR analysis was performed using a AVANCE DPX400 instrument (Bruker). 1H-NMR analysis was accomplished by dissolving the sample in DMSO-d6. 31P NMR spectroscopy was performed as per Argyropoulos et al. (1993). Fourier Transform Infrared (FTIR) spectra were recorded using a Thermo Nicolet 6700 instrument. Methoxyl groups in lignin were determined by subjecting the samples to acid hydrolysis (Ligero et al. 2008) and further HPLC determination of the methanol generated (using the same equipment and method cited above).
Manufacture of Lignin-PLA Composites and Mechanical Testing
Samples of lignin or esterified lignin were blended with PLA at 30:70 mass ratio using a co-rotating 16 mm twin screw Thermo PRISM EUROLAB 16 extruder with L/D ratio of 40. ISO 527 model 1A specimens for mechanical testing were produced by injection molding in a DEU 250H55 injection machine under the following conditions: injection nozzle temperature, 170 ºC; mold temperature, 25 ºC; injection speed, 10 mm/s; holding pressure, 20 bar; cooling time, 20 s. Tensile strength, stiffness and strain at break were determined in a Shimadzu Autograph AG-X testing machine (Shimadzu, Kyoto, Japan) with 50 kN load cell, operating at 2 mm·min−1crosshead speed.
RESULTS AND DISCUSSION
Recovery, Characterization, and Esterification of Lignin
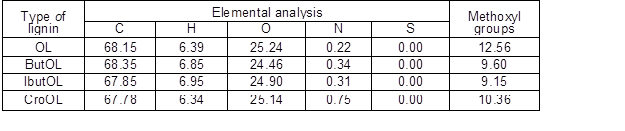
Table 2 lists data concerning the compositions of wood, pulp, and organosolv lignin recovered from the pulping liquor. Pine wood presented typical contents of ash and lignin, whereas the result reported for the glucan fraction accounted for the anhydroglucose units present in both cellulose and hemicelluloses (in this latter case, in the form of glucomannan). Hemicelluloses were mainly made up of mannose, with other monosaccharides appearing in lower proportions. Pulp was almost free of hemicelluloses, and was obtained at 42.8% yield (corresponding to 71.9% delignification); whereas the recovered organosolv lignin (OL) accounted for 87.3% of the dissolved lignin. The data in Table 1 confirm the high purity of OL, whereas the elemental analysis data (0.22 wt% nitrogen, 68.12 % carbon, 6.39% hydrogen, and 25.22% oxygen) indicated that the organosolv lignin was sulphur-free.
Methoxyl groups accounted for 12.56 wt% of OL, and the 31P-RMN spectra recorded for this material (see Fig. 1) showed that the hydroxyl groups were linked mainly to guayacil moieties and aliphatic chains, with minor proportions attached to syringyl, condensed and p-hydroxyphenil units.
The relative increase of lignin weight upon esterification (measured in terms of mass gain percentage, MGP) was employed to measure the extent of the reactions. According to the experimental data in Table 1, the reactions of organosolv lignin with butyric, isobutyric, or crotonic anhydrides (leading to esterified samples denoted ButOL, IbutOL and CroOL, respectively) resulted in MGP in the range 28.4 to 34.0. Table 3 lists compositional data for these materials. All samples showed total lignin contents (including the acid-soluble and acid-insoluble fractions) above 87 wt%; with low proportions of ash and carbohydrates. Most of the mass fraction not identified in ButOL, IbutOL, and CroEL corresponded to the contribution of ester groups, which were split under the analysis conditions.
Table 4 lists the results obtained in the elemental analysis of the various esterified lignins, as well as the ones concerning the methoxyl group contents. Closely related elemental compositions were determined for the various types of esterified lignins, which showed slightly decreased contents of oxygen and methoxyl groups respect to OL. In comparison, the H and C contents of OL were in the range determined for the modified lignins.
Table 2. Composition of Wood, Pulp, and Recovered Lignin (data in wt %)


Fig. 1. 31P-RMN spectra recorded for crude and derivatized lignins
Table 3. Composition of Esterified Lignins (data in wt%)

Table 4. Additional Analytical Data for OL and Esterified Lignins (wt%)

The 31P-NMR spectra recorded for lignins derivatized with butyric or isobutyric anhydrides (Fig. 1) did not present signals for hydroxyl groups, whereas the lignin derivatized with crotonic anhydride showed small signals corresponding to hydroxyls linked to aliphatic chains or guayacyl groups. On the other hand, the 1H-RMN data showed signals corresponding to ester groups generated upon derivatization with the various anhydrides. Besides the signals for aromatic H (8-6 ppm) and methoxyl H (3.8-3.5 ppm), other signals were ascribed as follows for the various types of materials:
- Butyrated lignin: -CH3, 1.1-0.7 ppm; β-CH2-, 1.8-1.4 ppm; α-CH2-, 2.4-2 ppm.
- Isobutyrated lignin: -CH3, 1.3-0.9 ppm; -CH-, 2.8-2.6 ppm.
- Crotonated lignin: CH3-, 2.1-1.7 ppm; vinylic H-, 6.2-6 ppm.
Figure 2 shows the FTIR spectra recorded for both OL and modified lignins. The fingerprint region (1600 to 600 cm-1) of the various samples presents differences caused by functionalization. Outside the fingerprint region, the strong wide bands between 3500 and 3100 cm-1 present in OL (corresponding to OH groups) did not appear in the spectra recorded for the modified lignins; whereas the bands between 2970 and 2840 cm-1 are ascribed to the stretching vibrations of C-H bonds, and the signal at 2843 cm-1 is ascribed to methoxyl group stretching. In comparison with the spectral data observed for OL, those recorded for ButOL and IbutOL exhibited larger bands at 2964 and 2934 cm-1, which are characteristic for methyl and methylene groups, respectively. As expected, esterified lignins presented weaker absorption bands corresponding to hydroxyl groups, and stronger absorption bands in the carbonyl group. The carbonyl absorption frequency depends on both spatial and structural factors, in particular ring stress or conjugation with double bonds. ButOL and IbutOL presented strong bands at 1756 cm-1(indicative of aromatic carbonyl groups) and at 1733 cm-1 (indicative of aliphatic carbonyl groups). Furthermore, increased absorption was observed between 1150 and 1050 cm-1 due to the formation of the ester groups. The carbonyl group band observed for CroOL appeared at a lower wavenumber (1724 cm-1) owing to conjugation with the double bond; whereas the two significant bands appearing at 1652 and 709 cm-1 corresponded to the double bond and to the trans C-H bond respect to the C=C plane.

Fig. 2. FTIR spectra recorded for crude and esterified lignins
Table 5 lists the 31P NMR results concerning the type and abundance of hydroxyl groups in OL and esterified lignins, which are in agreement with the structural features already discussed on the basis of the FTIR data. Almost complete esterification was observed for ButOL and IbutOL, with 97% decrease of aliphatic and aromatic -OH groups per C9 unit. In the case of CroOL, esterification was not complete: the amount of OH groups per C9 unit decreased by 78%; and the residual hydroxyl groups of aliphatic or aromatic nature accounted for 0.08 or 0.1 OH per C9 unit, respectively.
Thermal Analysis
Figure 3 shows the TGA and DTG curves determined for OL and esterified lignins under oxidative atmosphere. The total weight loss upon heating up to 200 ºC was below 5% in all the cases. The DTG data confirmed that the thermal decomposition took place in successive steps, with noticeable differences among samples. OL was decomposed along a broad temperature range, a fact ascribed to the existence of various oxygenated functional groups with different thermal stabilities. In comparison, the DTG profiles determined for esterified lignins presented more defined peaks, with at least three regions. The first region (temperatures in the range 20 to 110 ºC) showed a peak around 80 ºC caused by the evaporation of free or bonded water. In the second region (temperature range, 200 to 440 ºC), a defined peak was observed around 390 ºC, ascribed to the breaking of the bonds between the monomeric unit of lignin releasing the corresponding monomers (Vallejos et al. 2011). The third region (temperature range, 440 to 600 ºC) showed a peak around 560 ºC, ascribed to the decomposition of the aromatic ring. The temperature corresponding to the maximum decomposition rate was lower for OL (318 ºC) than for the esterifed lignins (386 to 396 ºC), whereas temperatures above 614 ºC resulted in formation of char residues at mass proportions below 3%. Decomposition was limited operating in nitrogen atmosphere (decomposed mass fractions: OL, 62%; ButOL, 68%; IbutOL, 70%; CroOL, 83%).
Table 5. Types and Abundance of Hydroxyl Groups in OL and Esterified Lignins, Determined by 31P-NMR



Fig. 3. TGA and DTGA curves recorded for crude and esterified lignins in air
Mechanical Testing
Table 6 lists the results obtained in the mechanical tests performed with samples made up of crude PLA or with various blends of PLA with OL or esterified lignins. The tensile strengths determined for PLA-OL blends were poorer than those obtained for neat PLA. Interestingly, some of the esterified lignins improved the plastic properties respect to PLA, as confirmed by their higher strains at break (Fig. 4). The suitability of ButEOL as a plasticizer was confirmed by its ability for increasing the strain at break of the corresponding blend up to 18.1% (in comparison with 1.88% for the reference material). IbutOL presented a limited ability as a plasticizer (strain at break, 5.40%), whereas no effects on this property were observed for PLA-CroOL blends. The tensile strength decreased by about 30% when PLA was blended with ButOL, IbutOL or
Table 6. Mechanical Properties of Crude PLA and Blends of PLA with OL or Esterified Lignins (data expressed as mean ± standard deviations)


Fig. 4. Stress-strain graphs for samples made up of crude PLA and blends of PLA with crude or esterified lignins
CroOL. Oppositely, the blends of PLA with esterified lignin showed higher Young’s modulus. This finding was especially noticeable for the blends PLA-ButOL and PLA-IbutOL, for which the Young’s modulus increased by 55 and 54%, respectively, to neat PLA, respectively. In comparison, PLA-IbutOL did not improve the modulus respect to the reference material
CONCLUSIONS
- Pinus pinaster wood samples were extensively delignified in ethanol/water media catalyzed with sulfuric acid operating under selected conditions.
- The lignin recovered from pulping presented high purity (with 95.7 wt % acid-insoluble lignin content), and was essentially free from sulfur.
- Lignin was reacted with three esterification agents (butyric, isobutyric, and crotonic anhydrides) for assessing the possible improvements in plastic properties. Esterification led to almost complete conversion of aliphatic and aromatic hydroxyl groups in samples derivatized with butyric or isobutyric anhydrides.
- Partial esterification was observed upon esterification with crotonic anhydride, a fact ascribed to the different reaction medium (dioxane instead the derivatization agent itself).
- The blends of PLA with esterified lignins showed better plastic properties than neat PLA; and the highest strain at break was recorded for blends of PLA with lignin esterified with butyric anhydride (a material that also presented a remarkably increased Young´s modulus respect to pure PLA).
ACKNOWLEDGMENTS
The authors are grateful to the Spanish “Ministry of Economy and Competitivity” for supporting this study in the framework of the research project “Advanced processing technonologies for biorefineries” (reference CTQ2014-53461-R), partially funded by the FEDER program of the European Union.
REFERENCES CITED
Argyropoulos, D. S., Bolker, H. I., Heitner, C., and Archipov, Y. (1993). “31P NMR spectroscopy in wood chemistry. Part V. Qualitative analysis of lignin functional groups,” J. Wood Chem. Technol. 13, 187-212. DOI:10.1080/02773819308020514.
Chung,Y. L., Olsson, J. V., Li, R. J., Frank, C. W., Waymouth, R. M., Billington, S. L., and Sattely, E. S. (2013). “A renewable lignin–lactide copolymer and application in biobased composites,” ACS Sust. Chem. Eng. 10, 1231-1238. DOI: 10.1021/sc4000835
Gordobil, O., Egüés, I., Llano-Ponte, R., and Labidi, J. (2014). “Physicochemical properties of PLA-lignin blends,” Polym. Degrad. Stabil. 108, 330-338. DOI: 10.1016/j.polymdegradstab.2014.01.002
Hilburg, S. L., Elder, A. N., Chung, H., Ferebee, R. L., Bockstaller, M. R., and Washburn, N. R. (2014). “A universal route towards thermoplastic lignin composites with improved mechanical properties,” Polymer 55, 995-1003. DOI: 10.1016/j.polymer.2013.12.070
Jeong, H., Park, J., Kim, S., Lee. J., and Cho, J. W. (2012). “Use of acetylated softwood kraft lignin as filler in synthetic polymers,” Fibers Polym. 13, 1310-1318. DOI: 10.1007/s12221-012-1310-6.
Li, J., He, Y., and Inoue, Y. (2003). “Thermal and mechanical properties of biodegradable blends of poly(L-lactic acid) and lignin,” Polymer Int. 52, 949-955. DOI: 10.1002/pi.1137
Ligero, P., Villaverde, J. J., de Vega, A., and Bao, M. (2008). “Delignification of Eucalyptus globulus saplings in two organosolv systems (formic and acetic acid). Preliminary analysis of dissolved lignins,” Ind. Crops Prod. 2, 110-117. DOI: 10.1016/j.indcrop.2007.08.008
Liu, H., and Zhang, J. (2011). “Research progress in toughening modification of poly(lactic acid),” J. Polym. Sci. Part B: Polym. Phys. 49, 1051-1083. DOI: 10.1002/polb.22283
Ouyang, W., Huang, Y., Luo, H., and Wang, D. (2012). “Poly(lactic acid) blended with cellulolytic enzyme lignin: Mechanical and thermal properties and morphology evaluation,” J. Polym. Environ. 20, 1-9. DOI 10.1007/s10924-011-0359-4.
Pérez-Guerrero, P., Lisperguer, J., Navarrete, J., and Rodriguez, D. (2014). “Effect of modified Eucalyptus nitens lignin on the morphology and thermo-mechanical properties of recycled polystyrene,” BioResources 9, 6514-6526. DOI: 10.15376/biores.9.4.6514-6526.
Spiridon, I., Leluk, K., Resmerita, A. M., and Darie, R. N. (2015). “Evaluation of PLA–lignin bioplastics properties before and after accelerated weathering,” Composites Part B: Engineering69, 342-349. DOI: 10.1016/j.compositesb.2014.10.006.
Thielemans, W., Can, E., Morye, S. S., and Wool, R. (2002). “Novel applications of lignin in composite materials,” J. Appl. Polym. Sci. 83, 323-331. DOI: 10.1002/app.2247.
Thielemans, W., and Wool, R. P. (2005). Lignin esters for use in unsaturated thermosets: lignin modification and solubility modeling. Biomacromolecules 6, 1895-1905. DOI: 10.1021/bm0500345
Toriz, G., Denes, F., and Young, R. A. (2002). “Lignin-polypropylene composites. Part 1: Composites from unmodified lignin and polypropylene,” Polym. Composites 23, 806-813. DOI: 10.1002/pc.10478
Vallejos, M. E., Felissia, F. E., Curvelo, A. A. S., Zambon, M. D., Ramos, L., and Area, M. C. (2011). “Chemical and physico-chemical characterization of lignins obtained from ethanol-water fractionation of bagasse,” BioResources 6, 1158-1171
Article submitted: March 1, 2016; Peer review completed: April 10, 2016; Revised version received and accepted: April 21, 2016; Published: April 28, 2016.
DOI: 10.15376/biores.11.2.5322-5332