
Abstract
Microcrystalline cellulose-based hydrogels were made using never-dried MCC (AaltoCellTM) as a raw material for a high-pressure mechanical treatment consisting of one to five passes at 700 bars. The effects of the mechanical treatment on the crystalline structure, morphology, geometrical dimensions, and specific surface area as well as rheological properties of the manufactured cellulose gel product were investigated. The results indicated that the process detached part of the crystalline area of the cellulose, resulting in loose particle architecture, increased surface area and porosity, and thus more accessible and reactive material. Due to the creation of the new internal surface area and porosity, more hydrogen bonds were formed between the cellulose particles, consequently creating more stable cellulose hydrogel-like slurries. The properties of the produced hydrogels were greatly influenced by the number of the treatment passes through the process equipment. Several passes through the process produced stronger cellulose hydrogels capable of retaining more water.
Download PDF
Full Article
Manufacturing of Hydrogels from Never-Dried Microcrystalline Cellulose by High-Pressure Mechanical Treatment
Annina Lähdeniemi,a,* Kari M. Vanhatalo,b and Olli Dahl a
Microcrystalline cellulose-based hydrogels were made using never-dried MCC (AaltoCellTM) as a raw material for a high-pressure mechanical treatment consisting of one to five passes at 700 bars. The effects of the mechanical treatment on the crystalline structure, morphology, geometrical dimensions, and specific surface area as well as rheological properties of the manufactured cellulose gel product were investigated. The results indicated that the process detached part of the crystalline area of the cellulose, resulting in loose particle architecture, increased surface area and porosity, and thus more accessible and reactive material. Due to the creation of the new internal surface area and porosity, more hydrogen bonds were formed between the cellulose particles, consequently creating more stable cellulose hydrogel-like slurries. The properties of the produced hydrogels were greatly influenced by the number of the treatment passes through the process equipment. Several passes through the process produced stronger cellulose hydrogels capable of retaining more water.
Keywords: Microcrystalline cellulose; MCC; Cellulose mechanical treatment; Dispersionizer; Cellulose hydrogel; Never-dried MCC; High-pressure treatment
Contact information: a: Department of Bioproducts and Biosystems, Aalto University School of Chemical Technology, PO Box 16300, Vuorimiehentie 1, Espoo, Finland; b: Andritz Oy, Tammasaarenkatu 1, 00180 Helsinki, Finland; *Corresponding author: annina.lahdeniemi@aalto.fi
INTRODUCTION
Developing new bio-based polymer nanomaterials for a wide range of applications has been recently emphasized in accordance with the increasing awareness of sustainability and environmental concerns. Natural cellulose-based materials are renewable, biodegradable, biocompatible, and non-toxic. Creating nano- and microscale cellulose products from wood cell walls using various different mechanical and chemical treatments, as well as their combinations, is the subject of significant research. The properties, functionality, durability, and uniformity required for the next generation of cellulose-based products and engineering applications cannot be achieved with traditional cellulosic products and materials.
Cellulose is the structural material of the fibrous cells; it has high levels of strength and stiffness per unit weight. The physical and morphological structures of cellulose are highly complex. The crystalline part of the cellulosic structure is dense and rigid due to the hydrogen bonding between molecular chains, which can explain its resistance against chemical and enzymatical treatments. The less ordered amorphous region of cellulose consists of polymer chains in a loose architecture that is easier to process (Klemm et al. 2005a; Himmel et al. 2007). Extracting cellulose to smaller scale, the majority of the defects associated with the hierarchical structure can be removed. Cellulose nanoparticles have high aspect ratio, low density, and a reactive surface of hydroxyl side groups that facilitates the grafting of the chemical properties to achieve custom-made surface properties. The accessibility and reactivity of cellulose materials are equal to the amount of free hydroxyl (-OH) groups on the surface of cellulose (Klemm et al. 2005b; Missoum et al. 2013). By creating new surface area and more porous material, free hydroxyl groups are available for reactions, and the processing of cellulose material becomes more effective.
Mechanical approaches to separate cellulosic fibers into nano- or microscale include refining and homogenizing, microfluidization, grinding, cryocrushing, and high intensity ultrasonication (Khalid et al. 2014). Devices such as a high-pressure homogenizer (HPH), Valley beater, Microfluidizer, ball mill, Masuko grinder, aqueous counter collision (ACC), and rotor stator cavitron have been tested (Manninen et al. 2011; Ankerfors 2012; Guezennec 2012; Taheri and Samyn 2012; Ardanuy et al. 2012; Kondo et al. 2014; Zhang et al. 2015). Mechanical processing produces high shear that causes transverse cleavage along the longitudinal axis of the cellulose microfibrillar structure, resulting in the extraction of long cellulose fibrils. The size reduction of cellulosic chains occurs due to the increased mechanical damage to the crystalline cellulose, resulting in decreased crystallinity (Moon et al. 2011; Lengkowski 2018).
In one of the first cellulose nanofiber (CNF) studies, Turbak et al. (1983) processed chemical pulp by using HPH, where the physical form of the used pulp changed from fiber form to a gel-like material. The high-pressure production equipment delaminates the cell walls of cellulose fibers, liberating the small-scale cellulose fibrils. When dispersed in water, these highly surface charged fibrils form a stable dispersed transparent or semi-opaque gel-like suspension, depending on the number of remaining larger particles. The aqueous suspension properties are influenced by hydrogen bonding between fibrils and water molecules and have rheological properties displaying partial gel-like structure.
Cellulose hydrogel is a polymeric material with highly networked structure that retains a large amount of water while maintaining its shape. Hydrogels are also hydrophilic materials that swell in water or aqueous solution without dissolution. At present, hydrogels are used in high value applications in different pharmaceutical products, as thickeners and stabilizers in food products and cosmetic formulations. Other existing and potential applications for cellulose gel materials include barrier films, antimicrobial films, transparent films, flexible displays, reinforcing fillers for polymers, biomedical implants, fibers and textiles, templates for electronic components, separation membranes, batteries, supercapacitors, and electroactive polymers, etc. (Moon et al. 2011).
Many attempts have been made to improve upon various properties of hydrogels, for example, enhanced strength, water content, permeability or biocompatibility to suit various applications. Often these improvements are achieved with mixed results. Typically, enhancing one property comes at the expense of another. Great challenges arise from the cost-performance conditions due to the increasing complexity of the hydrogel production methods. Hence, there remains a need for upgraded hydrogels with more desirable properties and for further development of more economical and sustainable methods for producing such fibrillary material.
The main drawbacks of producing hydrogels with high-pressure devices are clogging problems, caused by the very small orifice size, and the high process energy consumption. To overcome these disadvantages, it is favorable to reduce the size of the fibers before their passing through the equipment (Khalid et al. 2014). Hence, microcrystalline cellulose (MCC) consisting of bundles of crystallites with different particles has attracted attention as a promising raw material for the high-pressure equipment. Furthermore, MCC particles can immobilize high amounts of water on external and internal surfaces, which is accompanied by the formation of gel-like water systems (Ioelovich and Leykin 2008).
Yokota and Okumura (1984) were among the first to use MCC as a raw material to produce cellulose hydrogels. They dispersed MCC in water, which was homogenized using 560 bar feed pressure; after 20 passes a viscous gel was generated. Kleinebudde (2000) homogenized MCC dispersions with 500 bars and achieved a significant decrease in particle size distribution accompanied with an increase in the specific surface area (SSA). Jacquet et al. (2012) obtained a microfibril suspension from MCC with numerous (25 to 50) homogenization (500 bars) passes with significantly increased surface area. Lee et al. (2009) used a microfluidizer that disintegrated the particle form MCC to smaller fragments. Kondo et al. (2014) refined MCC with ACC with different number of passes resulting in 20 to 40x and 550 to 785x decrease for fiber length and width, respectively.
In the above-mentioned studies, the raw material was dry MCC because it has been the only commercially available MCC product. The difference in the properties of the dispersions manufactured from different MCC-types is neither clear nor much researched. Upon the drying of cellulose, the formation of internal hydrogen bonds between cellulose fibrils makes a secondary separation into single fibrils more difficult; this phenomenon is known as hornification (Diniz et al. 2004). Ioelovich and Leykin (2008) reported that the HPH-processed dispersions containing never-dried MCC particles had larger internal specific surface values and higher amounts of retained water compared to the ones with traditional dried-MCC. Drying the MCC causes decreases in the particles’ internal surface and the viscosity of the HPH-manufactured gel dispersions, which is explained by the irreversible closing of the internal MCC pores.
In this study, unmodified, never-dried microcrystalline cellulose MCC (AaltoCell™ MCC) as a raw material was mechanically processed with dispersionizer equipment using 700 bar homogenizing pressures to modify the structure of cellulose. The main aim was to discover a simple and efficient method for manufacturing cellulose hydrogels without the use of any additives (such as CMC). Due to the absence of additives and nano-size particles, the manufactured hydrogels should be highly applicable for food additives and drug delivery. The effects of the mechanical dispersionizer treatment on the crystalline structure, morphology, geometrical dimensions, rheology, and specific surface area of never-dried microcrystalline cellulose were investigated. The never-dried MCC was produced with AaltoCell™, a procedure that has a low chemical consumption and can thus produce lower cost MCC.
EXPERIMENTAL
Cellulose Raw Material and the Production of MCC (AaltoCell™)
Cellulose raw material (CelRaw) was never-dried microcrystalline cellulose manufactured with the AaltoCell™ method, as described in Vanhatalo and Dahl (2014). In brief, bleached softwood kraft pulp was hydrolyzed with a 1.5% dosage of sulfuric acid at 160 °C in 10% consistency for 110 min using tube like reactor with volume of 2.5 dm3. After the hydrolysis, the reactor was cooled, and the MCC material was washed with distilled water in a Büchner funnel until the washed filtrate conductivity was below 5.0 μS. The washed MCC material was centrifuged at 4500 rpm with a filter bag to the dry consistency of 45%.
Production of the Cellulose Hydrogels
The processing pressure in the dispersionizer was 700 bars, and the cellulose-water slurry temperature was kept under 80 °C during processing. Samples were collected after 1, 3, and 5 dispersionizer passes and stored at 4 °C.
CelRaw was diluted with distilled water to different consistencies—4.5%, 6.0%, 7.5%, 10.0%, 12.5%, and 15.0%—and samples were denoted as Cel-4.5, Cel-6.0, Cel-7.5, Cel-10.0, Cel-12.5, and Cel-15.0 respectively. These cellulose-water mixes were feed materials to Netzsch Omega® 60 Economic Dispersionizer equipment (Selb, Germany). This processing unit combines a nozzle and homogenizing valve technology and creates shear-, elongation-, turbulence-, cavitation-, and impact effects (Fig. 1).
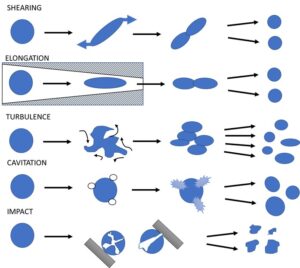
Fig. 1. Effects of the dispersionizer equipment (Source: Figure redrawn from the original one from a Netzsch brochure)
Test Standards and Methods
Particle size and WRV measurements were conducted for the wet samples. Other analyses were done for the dry samples prepared though a solvent exchange procedure with fully water miscible low molecular alcohol to avoid hornification or other structural changes that occur with the presence of water during cellulose drying. A solvent exchange procedure was completed with approximately 1.0 g of the sample (calculated as oven dry basis), which was measured into a 50-mL centrifugal tube. The tube was filled with tert-butanol, shaken vigorously, and centrifuged at 7500 rpm for 10 min. The liquid fraction was decanted, and the tube was filled again with tert-butanol. This procedure was repeated five times. The solvent exchanged cellulose samples were placed at -22 °C for 4 h. Finally, the samples were lyophilized with a freeze dryer (Labconco Freezone 2.5, Kansas City, USA) for 12 h.
The particle size distribution of the samples was measured with a Mastersizer 2000 equipped with a Hydro 2000MU dispersion unit (Malvern Instrument Ltd, Worcestershire, UK). The measurement was conducted as published (Vanhatalo and Dahl 2014). Particle size distribution d(0.1), d(0.5), and d(0.9) values were reported to describe physical particle sizes. All measurements were done in triplicate.
The ability to retain water was characterized by WRV measurement. About 5.0 g of the sample was measured into a plastic centrifuge tube equipped with 0.45 µm membrane filter (Macrosep Advance, Pall Corporation, Port Washington, USA). The sample was centrifuged at 3000 g for 5 and 20 min. The centrifuged and dewatered sample was weighed and oven-dried at 105 °C for 16 h. WRV was calculated as a ratio between the centrifuged wet sample and the oven dry sample. All measurements were performed in triplicate.
X-ray diffraction was performed on an X’Pert PRO MPD Alpha-1 diffractometer (PANalytical, Almelo, Netherlands) using Ge-filtered CuKα radiation beam generated at 45 kV and 40 mA. Scans were measured at 2θ in the range of 5 to 50. Crystallinity index values were calculated with two methods: amorphous subtraction and deconvolution methods (Park et al. 2010). In the amorphous subtraction method, alkali lignin was used to determine the amorphous background. The crystallinity index was calculated by Eq. 1 (Sathitsuksanoh et al. 2011),
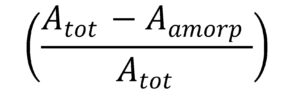
(1)
where Atot is area of sample diffractogram and Aamorp is area of amorphous diffractogram. In the deconvolution method, the individual crystalline peaks were extracted from diffractograms by using PeakFit 4.2 software (Systat Software Inc., San Jose, USA) assuming the Gaussian functions for each peak (Hult et al. 2003). Crystallinity index was calculated by dividing the area of crystalline peaks with the total area of the diffractogram. The d-spacing was calculated by Bragg’s equation, and the crystallite sizes were calculated from the widths of diffraction peaks using the Scherrer equation (Eq. 2),
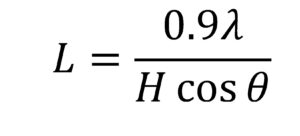
, (2)
where L is the crystallite size (nm) perpendicular to the plane, λ is the wavelength of X-ray (0.15418 nm), H is the full-width at half-maximum (FWHM) in radians, and θ is the Bragg angle.
Specific surface area (SSA), pore volume, and pore width were determined with the multi-point BET method (Brunauer et al. 1938). Pore size and volume were analyzed with the BJH method (Barrett et al. 1951) using a Micromeritics Tristar II 3020 (Micromeritics, Norcross, USA) that uses nitrogen gas as an adsorbate. Before the measurement, the samples were pretreated under nitrogen gas flow according to the following procedure: 15 min at 25 °C, 1 h at 60 °C, and 15 min at 25 °C. All measurements were performed in triplicate.
The distribution of molecular size (DM) and weight average molecular weight (Mw) were analyzed with gel permeation chromatography (GPC) carried out in a Dionex Ultimate 3000 system using a guard column (PLgel Mixed-A, 7.5 x 50 mm, Agilent Technologies, Santa Clara, USA), four analytical columns (PLgel Mixed-A, 7.5 x 300 mm) combined with a RI detection system (Shodex RI-101, Showa Denko K.K, Japan). Samples to the GPC measurement were prepared according to a published procedure (Testova et al. 2014). All measurements were conducted with double injection from each sample.
The structural morphology of the CelRaw and hydrogel samples were observed with scanning electron microscopy (Zeiss Ʃigma-VP, Jena, Germany) at 1.5 keV, equipped with an Everhart-Thornley detector. Prior to imaging, the samples were sputter coated (Emitech K-100X, Lewes, UK) with a 10-nm gold layer to obtain proper conductivity.
Settling tests were conducted as follows. CelRaw and hydrogel samples were diluted to 1.5% consistency and mixed 1 for min with an Ultra Turrax mixer (T25 IKA, Staufen im Breisgau, Germany). Dispersed, diluted gel mixtures were poured into transparent measuring cylinders to detect settling ability. Settling tests lasted 30 days. To examine and document the settling rate, photographs were taken intermittently.
Rheological investigations were performed using a strain-controlled rheometer (Physica MCR-300 Anton Paar, Ashland, VA, USA) equipped with 25-mm diameter surface-profiled (serrated) plate-to-plate geometrics. All cellulose hydrogels were measured at 23 °C using a 2-mm measurement gap. A 20 s pre-shear stage was performed at a shear rate of 10s-1, followed by a settling time of 180 s prior to the amplitude sweep. The amplitude sweep was performed using a constant angular frequency (1 s-1) at increasing strain amplitudes (0.01 to 500%). The moduli value (G’ and G’’) at a damping factor tan() = 1 was used to describe the gel strength of the samples, which can also be related to the strength of the spatial network.
RESULTS AND DISCUSSION
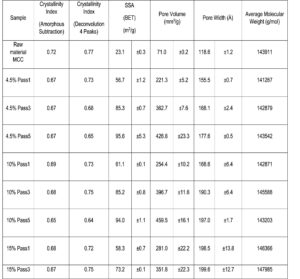
The crystallinity indexes, specific surface area (SSA), pore volume, pore width, and average molecular weight of the raw material and the treated cellulose hydrogel samples with different amount of passes through the mechanical treatment equipment are summarized in Table 1. The crystallinity of the gel form samples decreased with increasing dispersionizer passes with the larger consistency samples. However, after the first pass of the mechanical treatment, the crystallinity did not decrease much.
The results found in this study are similar to those reported previously (Ioelovich and Leykin 2008; Chen et al. 2010; Charani et al. 2013; Davoudpour et al. 2015; Lengkowski et al. 2018). Previous reports show that the crystallinity index of cellulose decreases with the increasing number of passes through similar high-pressure treatment. Lengkowski et al. (2018) claimed that as a result of this decrease in crystallinity, there are fewer intermolecular hydrogen bonds between the cellulose molecular chains. However, most of these studies were performed with different cellulose pulp raw material; only Ioelovich and Leykin (2008) used never-dried MCC as raw material.
Specific surface area (SSA) increased notably with every treatment pass, indicating enhanced accessibility of the gel-like state (Kleinebudde 2000; Ioelovich and Leykin 2008; Spence et al. 2011; Jacquet et al. 2012). In these studies, homogenized (dried and never-dried) MCC dispersions showed a significant increase in the specific surface area with numerous passes through the high-pressure equipment. A similar trend can be seen with the pore volume and pore width results of the cellulose hydrogel samples in Table 1. Only the pore width results were dependent on the dispersionizer feed consistency. A higher consistency was associated with a larger pore width.
Table 1. Raw Material and Cellulose Hydrogel Properties
The SEM images in Fig. 2 demonstrate the mechanical effect of the dispersionizer treatment to the cellulose particles. Even one pass through the process changed the cellulose architecture. The dense texture of the crystalline cellulose particles was broken into a looser, open web-like fiber structure with fibrillary surfaces. This action was reflected also in decreased CI values and particle sizes and increased SSA values.

Fig. 2. SEM of the CelRaw material (a and b), 4.5% cellulose gel after one pass (c and d) and five passes (e and f), 10% cellulose gel after five passes (g), and 15% consistency after five passes (h)
The web-like appearance of the treated cellulose gel samples is created by the numerous hydroxyl groups that are exposed throughout the cellulose particle chains (Lengkowski et al. 2018). The length of these chains can promote the formation of large areas of networks that guarantee numerous intermolecular hydrogen-bonding interactions between the nanofibers. Charani et al. (2013) also created similar cellulose gel slurries within which the microfibers were interconnected and formed an inherently entangled network.
Figure 2 shows that with the treatment of three to five passes through the dispersionizer, the forming of agglomerates decreased with the increased number of passes. There was no notable difference between the different consistency samples after the fifth pass. This result indicates that the optimal equipment setup for the treatment for such hydrogel samples in these conditions had been reached with considerably lower number of passes than, for example, as reported in Yokota and Kumura (1984).
Charani et al. (2013) proposed that the distribution of varying sizes of the pulp fiber particles in the cellulose hydrogels induces corresponding differences in the gel strength, making it an important factor in the rheology of the material suspensions and gels. Figure 3 shows the particle sizes of the treated cellulose hydrogel samples and the CelRaw material with the different particle size distributions. The particle size distribution d(0.1), which contains the smallest particles, did not exhibit any great change with any of the different cellulose hydrogel consistencies. Therefore, it was concluded that the dispersionizer process did not have any great effect on the smallest particles. This conclusion is also supported by the SEM images taken from the hydrogel samples that show no large amounts of small particle fragments.
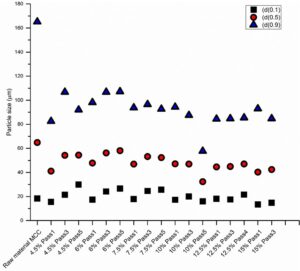
Fig. 3. Particle size d(0.1), d(0.5), and d(0.9) values for CelRaw and produced cellulose hydrogels
With the distribution d(0.5), the particle size was decreased by almost half during the first pass through the process when compared to the size of the same distribution of the CelRaw material. This decreasing action was similar with all treated samples consistencies with the distribution d(0.5). Compared with the other distributions, the dispersionizer process had greatest effect on the largest particles, which are represented by the particle size distribution d(0.9). Kleinebudde (2000) achieved a significant decrease in particle size distribution accompanied with an increase in liquid viscosity when homogenizing MCC dispersions, but not any true nanoscale fibers were observed in the produced cellulosic gel.
Considering the effect of the treatment on the side of the process consistency, the particles sizes of the lower consistency hydrogel samples (4.5%, 6%, and 7.5%) showed similar behavior with distributions d(0.1) and d(0.5) with first and third pass through the dispersionizer. With the higher consistency samples 6% and 7.5%, there were no major differences in any of the changes in all the distributions with the process passes 3 and 5. A similar trend was observed with the larger consistency samples (10%, 12.5%, and 15%). Thus, higher consistency was associated with a smaller effect on the particle size; the particle size increased more with smaller process consistencies. This may be due to the greater agglomeration or floc forming tendency of the MCC particles in the gel and/or the greater swelling tendency due to the larger amount of free water. Additionally, with the lower crystallinity index, the particles swell better due to the increased accessibility as well as the specific surface area (SSA).
With the process consistency hydrogel sample of 10%, the particle size decreased notably with the bigger size distributions d(0.5) and d(0.9). This behavior was dissimilar from the other treated consistency gel samples. These results may be related to the critical consistency of the floc forming tendency during the dispersionizer treatment.
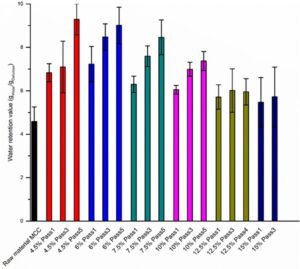
Fig. 4. WRV for CelRaw material MCC and produced cellulose hydrogels
The water retention value (WRV) demonstrates the water trapping nature of the treated MCC particles. Figure 4 represents the measured water retention values of the produced cellulose hydrogels. The WRV values increased visibly with the growing numbers of the treatment passes. Turbak et al. (1983) processed chemical pulp using HPH and noticed that as cellulose was homogenized further, the product had a higher water retention value. Zhang et al. (2012) and Spence et al. (2011) also produced cellulose gels with higher WRV values with Microfluidizer, HPH, and Masuko grinder.
In this study, the increase in WRV was greater with the smaller sample consistencies. The WRV values were larger with the smaller consistency samples. This may be the result of the greater swelling capacity of the lower consistency particles. The differences between the WRV values of the different passes were not notable with the larger hydrogel consistency samples 12.5% and 15%. There was a similar phenomenon with these particular samples in the particle size distributions. The biggest differences between the different sample consistencies was noted with the WRV values of the passes 3 and 5. Evidently, the ability of the cellulose hydrogels to hold water increased with the growing specific surface area SSA and pore volume and also with the decreasing particle sizes.
Figure 5 shows that there was no or very slight particle settling with the samples of the smallest consistency (4.5%). These samples formed an almost stable gel, indicating strong forces between the cellulose particles in the hydrogel slurry. This result is supported by the WRV values that are largest with this consistency in Fig. 4.

Fig. 5. Settling of the cellulose particles (%) as a function of time (min). Different samples are presented in the graphs: a), b) and c) cellulose hydrogels in 4.5% consistency after 1, 3, and 5 passes, respectively; d), e) and f) in 10% consistency after 1, 3, and 5 passes, respectively; g), h) in 15% consistency after 1 and 3 passes, respectively; and i) CelRaw material MCC.
With the larger consistency hydrogel samples, a larger number of passes through the dispersionizer stabilized the cellulose hydrogels, possibly due to the higher number of hydrogen bonding between the cellulose particles. With the treated samples, the settling occurred later and less vigorously. The higher consistency samples contained more cellulose particles, and they settled more rapidly. However, all samples formed relatively stable hydrogel slurries for about 6 h, compared with the untreated MCC slurry settling within min. This phenomenon and the effect of the process to the settling rate are shown in Fig. 6, where different hydrogel samples were allowed to settle over 2 days.
Rheology reflects the behavior of the cellulose suspensions in different industrial processes, such as mixing and pumping. Rheological procedures focusing on the viscoelastic properties are used to monitor the state of dispersion, the binding behavior of the cellulose surface for water, and the forming of the gel-like structure. Viscoelasticity reflects the gel-like material’s colloidal structure and its breakdown. Therefore, the alterations in the degree of particle entanglement, agglomeration, and/or flocculation in the network structure, are shown as a change of the mechanical and viscoelastic properties of the suspension, including gelation (Dimic-Misic et al 2018).

Fig. 6. Photograph illustrating the settling of the different cellulose hydrogel samples after 2 days 18 h, and 28 min
The gelation properties of the produced hydrogels were studied with moduli values G’ and G’’, which are a measure of the elastic component and a measure of the viscous component, respectively. These parameters describe the strength and behavior of the hydrogels, and they give indication of how fluid-like the produced gel material is. Viscoelastic storage (G’) and loss modulus (G’’), are illustrated in Figs. 7 through 9 as functions of the shear strain of the manufactured cellulose hydrogels based on the results from strain sweep tests.
All of the processed hydrogel samples had a short linear viscoelastic region, which lengthened with the growing number of passes through the dispersionizer, exhibited as the more dispersed and gel-like suspension. This progress reflects the increased surface area and the greater number of thinner fibrils with more trapping water, leading to a strong homogeneous gel-like structure of the suspension. Linear viscoelastic region resembles the area of the curve where G’ remains constant as the more energy is applied to the sample. As the amplitude continues to increase, the structure of the material begins to break down. The strain where this breakdown occurs is called critical strain. Critical strain defines the end of the linear viscoelastic region and thus provides information on the nature of the internal structure of the gel material.
The most important references for this study are Dimic-Misic et al. (2018) and Dong et al. (2018), as both of them used the same raw material: never-dried MCC made with the AaltoCellTM procedure. These studies are in good accordance with each other. All of the researched hydrogel samples showed a constant modulus until the stress passed the yield point of the gel and decreased remarkably. The values of G’’ were typically a 10-fold less to the G’ values for each hydrogel samples.
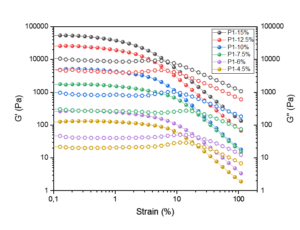
Fig. 7. The storage modulus, G’ (filled dots) and the loss modulus, G’’ (unfilled dots) as a function of the shear strain of the manufactured cellulose gels after one pass through the dispersionizer
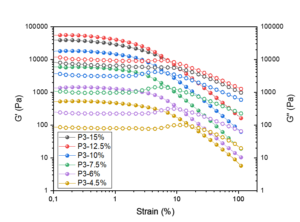
Fig. 8. The storage modulus, G’ (filled dots) and the loss modulus, G’’ (unfilled dots) as a function of the shear strain of the manufactured cellulose gels after 3 passes through the homogenizer
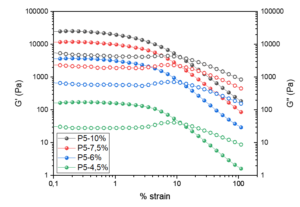
Fig. 9. The storage modulus, G’ (filled dots) and the loss modulus, G’’ (unfilled dots) as a function of the shear strain of the manufactured cellulose gels after five passes through the homogenizer
Dimic-Misic et al. (2018) explained that this difference in the modulae magnitudes reflects the combination of high colloidal stability derived from mutual charge repellence in addition to the combination of the surface adsorbed and trapped water structure. Furthermore, the magnitude of the modulae increased as a function of consistency increase.
The critical strain of the manufactured hydrogels increased with the number of passes through the dispersionizer. However, the critical strain appeared to be independent of the produced hydrogel concentration, as noted by Charani et al. (2013). The G’ results showed that the point of critical strain comes earlier and profounder with the higher cellulose hydrogel consistencies. Thus, less energy was required to break down the internal network structure of the more concentrated cellulose gel material. This effect was more apparent with the higher number of passes through the process.
In agreement with previous high-pressure treatment studies (Pääkkö et al. 2007; Iotti et al. 2011; Charani et al. 2013; Dimic-Misic et al. 2018; Dong et al 2018), both G’ and G’’ increased with higher cellulose hydrogel consistency. The hydrogels with higher consistencies produced more concentrated gels that subsequently led to the increase in modulae. This result indicates that these higher consistency cellulose hydrogels are more resilient, thus having higher storage and loss modulus.
The WRV values (shown in Fig. 4) increased along with the strength of the produced cellulose hydrogels. Thus, additional passes through the dispersionizer process produced stronger gels capable of retaining more water. This effect was less apparent with higher feed consistencies (6% to 10%). Stronger hydrogels were created due to the formation of new internal surface area and porosity. Dimic-Misic et al. (2018) claimed that the stronger gel-like behavior is produced by the more pronounced hydrogen bonding within the cellulose particle suspension due to the high surface area in the dispersed wet state. Vanhatalo (2017) proposed that the increased modulae and WRV values possibly result from the AaltoCell™-type MCC’s never-dried and thus more porous structures. This is also supported by the claim of Ioelovich and Leykin (2008) that the drying of MCC causes the irreversible closing of the particles’ internal pores. Never-dried MCC’s fibril-like structure enables the mechanical work to be applied directly to the particles as a function of specific surface. Thus using unnecessary energy for overcoming the mechanical elastic structure associated with entangled and retained fibers is avoided (Dimic-Misic et al. 2018). The potential of the use of never-dried MCC was described by Dong et al. (2018). The greater gel forming ability of the mechanically treated never-dried MCC particles compared with the dried commercially grade MCC is due to the opened fibrillar structure and higher elasticity of the never-dried material.
CONCLUSIONS
Even one treatment pass of never-dried microcrystalline cellulose (MCC) through the dispersionizer equipment removed part of the crystalline area of the cellulose, making the particle structure more accessible and porous. This increased its specific surface area and consequently the network swelling capacity. In addition, more hydrogen bonds were formed between the particles, creating more stable cellulose hydrogel.
The mechanical dispersionizer processing changed the structure of microcrystalline cellulose particles in macro as well as in molecule level, resulting in loosened fibrillar-type architecture, increased surface area, and thus more reactive cellulose material. This effect was greatest with the largest MCC particles.
The resulting damage from the dispersionizer process in the fiber structure generated a progressive increase in gelation of the hydrogel suspension. The higher consistency of pulp suspensions produced hydrogels with higher storage and loss modulus. Several passes through the process produced stronger cellulose hydrogels capable of retaining more water due to the formation of new internal surface area and porosity.
Processing never-dried MCC (produced with the AaltoCell™ procedure) with dispersionizer offers higher capacity production rate compared to the combination of the traditional high-pressure methods and commercial dried-mcc raw materials with much higher feed consistency (e.g., 1% vs. 10%), lower used pressure (e.g., 700 bar vs. 2000 bar) and fewer number of passes through the process. Higher consistency decreases transportation costs and increases the future processability of the hydrogels. Furthermore, during storage, the less aqueous environment inhibits the growth of undesirable micro-organisms.
This processing method allows the use of cellulose gel material in high-value-applications, such as food and pharmaceutical industry, due to its micro scale, international E-code status and no need of the additional chemicals in the mechanical treatment.
ACKNOWLEDGMENTS
The authors are grateful for the all of support given by the staff of Department of Bioproducts and Biosystems, Aalto University School of Chemical Technology.
REFERENCES CITED
Ankerfors, M. (2012). Microfibrillated Cellulose: Energy-efficient Preparation Techniques and Key Properties, Licentiate Thesis, KTH Royal Institute of Technology, Sweden
Ardanuy, M., Claramunt, J., Arévalo, R., Parés, F., Aracri, E., and Vidal T. (2012). “Nanofibrillated cellulose (NFC) as a potential reinforcement for high performance cement mortar composites,” BioResources 7(3), 3883-3894. DOI: 10.15376/biores.7.3.3883-3894
Barrett, E. P., Joyner, L. G., and Halenda, P. P. (1951). “The determination of pore volume and area distributions in porous substances. I, Computations from nitrogen isotherms,” Journal of the American Chemical Society 73, 373-380. DOI: 10.1021/ja01145a126
Brunauer, S., Emmett, P. H., and Teller, E. (1938). “Adsorption of gases in multimolecular layers,” Journal of the American Chemical Society 60, 309-319. DOI: 10.1021/ja01269a023
Charani, P. R., Dehghani-Firouzabadi, M., Afra, E., and Shakeri, A. (2013). “Rheological characterization of high concentrated MFC gel from kenaf unbleached pulp,” Cellulose 20, 727-740. DOI: 10.1007/s10570-013-9862-1
Chen, D., Guo, Y., Huang, R., Lu, Q., and Huang, J. (2010). “Pretreatment by ultra-high pressure explosion with homogenizer facilitates cellulase digestion of sugarcane bagasses,” Bioresource Technology 101, 5592-5600. DOI: 10.1016/j.biortech.2010.02.003
Davoudpour, Y., Hossain, S., Khalil, H. P. S. A., Haafiz, M. K. M., Ishak, Z. A. M., Hassan, A., and Sarker, Z. I. (2015). “Optimization of high-pressure homogenization parameters for the isolation of cellulosic nanofibers using response surface methodology,” Industrial Crops and Products 74, 381-387. DOI: 10.1016/j.indcrop.2015.05.029
Dimic-Misic, K., Vanhatalo, K., Dahl, O., and Gane, P. (2018). “Rheological properties comparison of aqueous dispersed nanocellulose derived from a novel pathway-produced microcrystalline cellulose or by conventional methods,” Applied Rheology 28(6), 64474. DOI: 10.3933/ApplRheol-28-64474
Diniz, F., Gil, M., and Castro, J. (2004). “Hornification – Its origin and interpretation in wood pulps,” Wood Science Technology 37, 489-494. DOI: 10.1007/s00226-003-0216-2
Dong, Y., Paukkonen, H., Fang, W., Kontturi, E., Laaksonen, T., and Laaksonen, P. (2018). “Entangled and colloidally stable microcrystalline cellulose matrices in controlled drug release,” International Journal of Pharmaceutics 548(1), 113-119. DOI: 10.1016/j.ijpharm.2018.06.022
Guezennec, C. (2012). Development of New Packing Materials Based on Micro- and Nano-fibrillated Cellulose, Ph.D. Dissertation, Grenoble University, France
Himmel, M. E., Ding, S. Y., Johnson, D. K., Adney, W. S., Nimlos, M. R., Brady, J. W., and Foust, T. D. (2007). “Biomass recalcitrance: Engineering plants and enzymes for biofuels production,” Science 315, 804-807. DOI: 10.1126/science.1137016
Hult, E. L., Iversen, T., and Sugiyama, J. (2003). “Characterization of the supermolecular structure of cellulose in wood pulp fibres,” Cellulose 10, 103-110. DOI: 10.1023/A:1024080700873
Ioelovich, M., and Leykin, A. (2008). “Study of solid-liquid interface in water dispersions of microcrystalline celluloses,” Composite Interfaces 15, 7-9, 771-779.
Jacquet, N., Vanderghem, C., Danthine, S., Blecker, C., and Paquet, M. (2012). “Influence of homogenization treatment on physicochemical properties and enzymatic hydrolysis rate of pure cellulose fibers,” Applied Biochemistry and Biotechnology 169, 1315-1328. DOI: 10.1007/s12010-012-0057-2
Kleinebudde, P. (2000). “Influence of degree of polymerization on behavior of cellulose during homogenization and extrusion/spheronization,” American Association of Pharmaceutical Scientists 2, 1-10. DOI: 10.1208/ps020321
Klemm, D., Heublein, B., Fink, H. P., and Bohn, A. (2005a). “Cellulose: Fascinating biopolymer and sustainable raw material,” Angewandte Chemie International Edition 44, 3358-3393. DOI: 10.1002/anie.200460587
Klemm, D., Schmauder, H-P., and Heinze, T. (2005b). “Cellulose,” in: Biopolymers Online, Wiley-VCH Verlag GmbH & Co. KGaA. DOI: 10.1002/3527600035.bpol6010
Kondo, T., Kose, R., Naito, H., and Kasai, W. (2014). “Aqueous counter collision using paired water jets as a novel means of preparing bio-nanofibers,” Carbohydrate Polymers 112, 284-290. DOI: 10.1016/j.carbpol.2014.05.064
Lee, S-Y., Chun, S-J., Kang, I-A., and Park, J-Y. (2009). “Preparation of cellulose nanofibrils by high-pressure homogenizer and cellulose-based composite films,” Journal of Industrial and Engineering Chemistry 15, 50-55. DOI: 10.1016/j.jiec.2008.07.008
Manninen, M., Kajanto, I., Happonen, J., and Paltakari, J. (2011). “The effect of microfibrillated cellulose addition on drying shrinkage and dimensional stability of wood-free paper,” Nordic Pulp and Paper Research Journal 26, 297-305. DOI: 10.3183/npprj-2011-26-03-p297-305
Missoum, K., Belgacem, M. N., and Bras, J. (2013). “Nanofibrillated cellulose surface modification: A review,” Materials 6, 1745-1766. DOI: 10.3390/ma6051745
Moon, R. J., Martini, A., Nairn, J., Simonsen, J., and Youngblood, J. (2011). “Cellulose nanomaterials review: Structure, properties and nanocomposites,” Chemical Society Reviews 40, 3941-3994. DOI: 10.1039/C0CS00108B
Park, S., Baker, J. O., Himmel, M. E., Parilla, P. A., and Johnson, D. K. (2010). “Cellulose crystallinity index: Measurement techniques and their impact on interpreting cellulase performance,” Biotechnology for Biofuels 3, 10. DOI: 10.1186/1754-6834-3-10
Pääkkö, M., Ankerfors, M., Kosonen, H., Nykänen, A., Ahola, S., Österberg, M., Ruokolainen, J., Laine, J., Larsson, P. T., Ilkkala, O., and Lindström, T. (2007). “Enzymatic hydrolysis combined with mechanical shearing and high-pressure homogenization for nanoscale cellulose fibrils and strong gels,” Biomacromolecules 8(6), 1934-41. DOI: 10.1021/bm061215p
Sathitsuksanoh, N., Zhu, Z. G., Wi, S., and Zhang, Y. H. P. (2011). “Cellulose solvent-based biomass pretreatment breaks highly ordered hydrogen bonds in cellulose fibers of switchgrass,” Biotechnology and Bioengineering 108, 521-529. DOI: 10.1002/bit.22964
Siqueira, G., Bras, J., and Dufresne, A. (2010). “New process of chemical grafting of cellulose nanoparticles with a long chain isocyanate,” Langmuir 26, 402-411. DOI: 10.1021/la9028595
Spence, K. L., Venditti, R. A., Rojas, O. J., Habibi, Y., and Pawlak, J. J. (2011). “A comparative study of energy consumption and physical properties of microfibrillated cellulose produced by different processing methods,” Cellulose 18, 1097-1111. DOI: 10.1007/s10570-011-9533-z
Testova, L., Borrega, M., Tolonen, L. K., Penttila, P. A., Serimaa, R., Larsson, P. T., and Sixta, H. (2014). “Dissolving-grade birch pulps produced under various prehydrolysis intensities: Quality, structure and applications,” Cellulose 21, 2007-2021. DOI: 10.1007/s10570-014-0182-x
Taheri, H., and Samyn, P. (2016). “Effect of homogenization (microfluidization) process parameters in mechanical production of micro- and nanofibrillated cellulose on its rheological and morphological properties,” Cellulose 23, 1221-1238. DOI: 10.1007/s10570-016-0866-5
Turbak, A. F., Snyder, F. W., and Sandberg, K. R. (1983). “Microfibrillated cellulose, a new cellulose product: Properties, uses, and commercial potential,” Journal of Applied Polymer Science, Applied Polymer Symposia 37, 815-828.
Yokota, H., and Okumura, Y. (1984). “Homogenization of microcrystalline cellulose suspension,” Japan patent JP 59120638.
Vanhatalo, K. M., and Dahl, O. P. (2014). “Effect of mild acid hydrolysis parameters on properties of microcrystalline cellulose,” BioResources 9(3), 4729-4740. DOI: 10.15376/biores.9.3.4729-4740
Vanhatalo, K. (2017). A New Manufacturing Process for Microcrystalline Cellulose (MCC), Ph.D. Dissertation, Department of Bioproducts and Biosystems, Aalto University, Espoo, Finland.
Zhang, L., Tsuzuki, T., and Wang, X. (2015). “Preparation of cellulose nanofiber from softwood pulp by ball milling,” Cellulose 22, 1729-1741. DOI: 10.1007/s10570-015-0582-6
Article submitted: March 17, 2020; Peer review completed: May 24, 2020; Revised version received and accepted: June 9, 2020; Published: February 1, 2021.
DOI: 10.15376/biores.16.2.2215-2234