
Abstract
Sulphate removal from mine water is a problem because traditional chemical precipitation does not remove all sulphates. In addition, it creates lime sediment as a secondary waste. Therefore, an inexpensive and environmental-friendly sulphate removal method is needed in addition to precipitation. In this study, carbon residues from a wood gasification process were repurposed as precursors to a suitable sorbent for SO42- ion removal. The raw material was modified using ZnCl2, BaCl2, CaCl2, FeCl3, or FeCl2. Carbon residues modified with FeCl3 were selected for further consideration because the removal efficiency toward sulphate was the highest. Batch sorption experiments were performed to evaluate the effects of the initial pH, initial SO42- ion concentration, and contact time on sulphate removal. The removal of SO42- ions using Fe-modified carbon residue was notably higher compared with unmodified carbon residue and commercially available activated carbon. The sorption data exhibited pseudo-second-order kinetics. The isotherm analysis indicated that the sorption data of Fe-modified carbon residues can be represented by the bi-Langmuir isotherm model.
Download PDF
Full Article
Sulphate Removal from Water by Carbon Residue from Biomass Gasification: Effect of Chemical Modification Methods on Sulphate Removal Efficiency
Hanna Runtti,a Sari Tuomikoski,*,a Teija Kangas,a Toivo Kuokkanen,a Jaakko Rämö, a and Ulla Lassi a, b
Sulphate removal from mine water is a problem because traditional chemical precipitation does not remove all sulphates. In addition, it creates lime sediment as a secondary waste. Therefore, an inexpensive and environmental-friendly sulphate removal method is needed in addition to precipitation. In this study, carbon residues from a wood gasification process were repurposed as precursors to a suitable sorbent for SO42- ion removal. The raw material was modified using ZnCl2, BaCl2, CaCl2, FeCl3, or FeCl2. Carbon residues modified with FeCl3 were selected for further consideration because the removal efficiency toward sulphate was the highest. Batch sorption experiments were performed to evaluate the effects of the initial pH, initial SO42- ion concentration, and contact time on sulphate removal. The removal of SO42- ions using Fe-modified carbon residue was notably higher compared with unmodified carbon residue and commercially available activated carbon. The sorption data exhibited pseudo-second-order kinetics. The isotherm analysis indicated that the sorption data of Fe-modified carbon residues can be represented by the bi-Langmuir isotherm model.
Keywords: Sorption; Sulphate; Chemical modification; Carbon residue; Activated carbon
Contact information: a: Research Unit of Sustainable Chemistry, University of Oulu, P.O. Box 3000, FI-90014 University of Oulu, Finland; b: Kokkola University Consortium Chydenius, Unit of Applied Chemistry, University of Jyvaskyla, Talonpojankatu 2B, FI-67100, Kokkola, Finland; *Corresponding author: sari.tuomikoski@oulu.fi
INTRODUCTION
Sulphate (SO42-) is a major pollutant that occurs in both natural waterways and industrial effluents, such as inorganic chemical industry wastewater and acid mine drainage. Sulphate is typically present in high concentrations, especially in industrial wastewaters, which may contain several thousand milligrams of SO42- ions per liter. The main natural sources of SO42- ions are chemical weathering and dissolution of sulphur-containing minerals (Rayner-Canham 1999; Cao et al. 2011; Rui et al. 2011). Excess SO42- ions cause an imbalance in the natural sulphur cycle and endanger human health. While SO42- ions are common in drinking water (Rui et al. 2011), many countries have not set guidelines because sulphate is less toxic than heavy metals (Silva et al. 2012). Sulphate ions are corrosive to reinforced steel (Rui et al. 2011). In Finland, the SO42- ion limit in drinking water is set at 250 mg/L, but it is recommended that the concentration not be higher than 150 mg/L to ensure that water pipes are not damaged (MSAH 2000). Sulphate concentrations higher than 600 mg/L can cause a laxative effect and affect the taste of the water (Silva et al. 2012). Typically, domestic sewage contains less than 500 mg/L of SO42- ions; therefore, the removal of sulphate with a large concentration range is a considerable task. Environmental agencies in many countries have set maximum SO42- ion values of 250 to 1000 mg/L in both mine drainage and industrial effluents (Namasivayam and Sureshkumar 2007; Silva et al. 2012).
Some established methods for the removal of SO42- ions are chemical precipitation, biological treatment, ion exchange, reverse osmosis, electrodialysis, and adsorption. Traditional chemical precipitation by barium or calcium salts or by lime does not remove all sulphates from water, and in addition, it creates sediment as a secondary waste (Silva et al. 2012). Low concentrations cannot be removed by lime precipitation because of the high solubility of the CaSO4 that is produced (Hartinger 1994). Therefore, an additional inexpensive and environmental-friendly sulphate removal method is required to supplement the precipitation process. An adsorption system has the potential to be used in a so-called hybrid-system with precipitation, in which the remaining sulphate concentrations after the precipitation process could be removed via adsorption.
The main advantages of biological sulphate reduction are the low volumes of sludge produced and the recovery of base metals as sulphides. On the other hand, biological treatment and ion exchange are expensive. Because of the high cost associated with chemical treatment technologies, and the long residence time requirement for biological treatment, there has been a concerted effort towards developing other sulphate removal technologies. Adsorption may be preferred to sulphate removal because it is rapid and its selectivity is high. In addition, sulphate can be recovered during adsorption. (EC 2008; Madzivire et al. 2010; Rui et al. 2011)
Sulphates can be adsorbed by γ-Al2O3 (Wu et al. 2002), chitin-based shrimp shells (Moret and Rubio 2003), coconut coir pith (Namasivayam and Sangeetha 2008), poly-m-phenylenediamine (Sang et al.2013), and goethite (Geelhoed et al. 1997). These adsorbents are modified by chemicals, such as BaCl2, ZrO(OH)2, and CaCl2, but these substances are expensive. Furthermore, methods using ZnCl2activation (Namasivayam and Sangeetha 2008), NaOH treatment (Moret and Rubio 2003), ammonium persulphate (Sang et al. 2013), cationic surfactants (Rui et al. 2011), and zirconium oxychloride (Mulinari and da Silva 2008) have been used to produce sorbents for SO42- ion removal. However, there is still a need for suitable sorbents that are produced on a large scale using environmental-friendly and inexpensive chemicals.
Typically, adsorption processes utilize commercial activated carbon (AC) to remove pollutants from wastewater. However, the high cost of AC sometimes deters its widespread use for wastewater treatment, and therefore, there is a need for alternative raw materials to make sorbents. In this study, the raw material was sourced from an energy generation process, i.e., biomass gasification in which carbonaceous material is formed as a waste material. Gasification is an effective technology that converts organic matter to heat and power and utilization of biomass. The amount of carbon residue (CR) is approximately 0.1 to 15% of the gasified material, and rather high volumes are produced (Knoef 2005). Because this high-carbon residue is classified as a waste material within the European Union, all utilization applications are preferred (EC 2008). Therefore, the utilization of this material is important for improving the cost-effectiveness of the gasification process. In addition, carbon residue includes metals, which could influence the adsorption activity of the residue (Kilpimaa et al. 2013).
In this study, CR was modified to remove SO42- ions from an aqueous solution. CR was chemically treated with different water-soluble inorganic compounds (ZnCl2, CaCl2, BaCl2, FeCl3, FeCl2), and the sorbent with the most efficient SO42- ion removal was selected for further consideration. Unmodified CR and commercial powdery activated carbon were used as reference materials. Batch experiments examined the influence of initial pH, initial SO42- ion concentration, and contact time on SO42- removal. In addition, the adsorption kinetics were modeled with pseudo-first-order and pseudo-second-order equations. Isotherm analysis was achieved using nonlinear Langmuir, nonlinear Freundlich, Sips, and bi-Langmuir isotherm models.
EXPERIMENTAL
Materials
Carbon residues obtained from a biomass gasification pilot plant (Sievi, Finland) were used as the raw material for the preparation of sorbents. A downdraft gasifier (150 kW) operating at 1000 °C, and wood chips (pine and spruce), were used as the raw materials for fuel, at a feeding rate of 50 kg/h. Powdered, commercially activated carbon (pro-analysis quality) was the reference sample (Norit, Zaandam, The Netherlands). To ensure uniformity and product quality, all materials were dried overnight at 110 °C, crushed, and sieved to obtain a particle diameter of less than 150 µm before use. Each modified product was also sieved to ensure particle sizes smaller than 150 µm.
Acid Washing
To reduce the ash content by selectively removing mineral components, CR was washed with a 1 N solution of HCl and H2SO4 (1:1) for 24 to 29 h at a liquid-to-solid (L/S) ratio of 10 (w/w). The CR samples were filtered and washed with distilled water using an L/S ratio of 40 (w/w) for at least 1 h. The samples were then filtered and dried at 110 °C overnight before being crushed and sieved to achieve particles of uniform quality (< 150 µm).
Chemical Activation
Chemical activation was performed using a wet impregnation method (Ahmadpour and Do 1996). First, CR was mixed with 5 M ZnCl2 (ACR 1), 1 M BaCl2 (ACR 2), or 1 M CaCl2 (ACR 3) at an L/S ratio of 10 (w/w). For ZnCl2, the contact time was 1 h, while for BaCl2 and CaCl2 solutions, the contact time was 5 min. Previously, the optimum ZnCl2 treatment was determined to be 1 h at 5 M concentration (Kilpimaa et al. 2014). The BaCl2 and CaCl2 solutions were not studied before, so a 1 M solution was used for 5 min. The mixture containing CR and chemical activating agent was dried at 110 °C overnight and activated at 500 °C for 1 h. The resulting products were washed sequentially with 0.5 M HCl for 10 min using an L/S of 10, hot distilled water, and finally, cold distilled water to remove residual chemicals or organic and mineral contents. Finally, the products were dried at 110 °C overnight, crushed, and sieved.
Chemical Modification
Protocol 1: 1 M BaCl2 (BaCl2∙2H2O) and 1 M CaCl2
CR was mixed with 1 M barium chloride, BaCl2∙2H2O (MCR 1), or calcium chloride, CaCl2 (MCR 2), with an L/S ratio of 10 (w/w). The mixture was shaken with a magnetic stirrer at room temperature for 18 h. The mixture was then filtered, washed carefully with distilled water, dried at 110 °C overnight, crushed, and sieved.
Protocol 2: 1 M FeCl2∙4H2O
Acid-washed CR was mixed with 1.0 M ferrous chloride, FeCl2∙4H2O (MCR 3), without headspace to prevent ferrous oxidation and precipitation (Chang et al. 2010). The mixture was shaken with a magnetic stirrer at room temperature for 24 h. The CR was then separated from the ferrous solution and dried overnight at 110 °C. During the post-treatment, Fe-impregnated CR was mixed with 1 M NaOH for 24 h and then soaked in 0.1 M HCl for 24 h to remove residual hydroxide. Finally, the Fe-impregnated CR was washed thoroughly using distilled water, dried at 110 °C, crushed, and sieved.
Protocol 3: 1 M FeCl3
Fe-impregnated CR (MCR 4) was prepared by mixing acid-washed CR with 1 M ferric chloride (FeCl3) at an L/S ratio of 10 (w/w). The mixture was shaken with a magnetic stirrer at room temperature for 24 h. The solution was dried at 110 °C for 24 h, washed with distilled water, dried again at 110 °C, crushed, and sieved.
Protocol 4: 1 M FeCl3
Fe-impregnated CR (MCR 5) was prepared as in Protocol 3. The only difference between Protocols 3 and 4 was the raw material. In Protocol 3, the raw material was acid washed, but in Protocol 4, the modification was performed without acid washing.
Protocol 5: 2 M FeCl3
Fe-impregnated CR (MCR 6) was generated by mixing the raw material (without acid washing) with 2 M FeCl3 at an L/S ratio of 4 (w/w). Diluted NaOH was added to obtain a pH of approximately 4. The solution was then dried at 110 °C, washed with 1 L of distilled water, dried again at 110 °C, crushed, and sieved (Chen et al. 2007).
Characterization Methods
The specific surface area of the products were determined from nitrogen adsorption–desorption isotherms at the temperature of liquid nitrogen (-196 °C), using a Micromeritics ASAP 2020 (Norcross, GA, USA). The same equipment was used to determine the pore size and pore volume.
The carbon content (TC) of the CR and AC were determined by elementary analysis using a Flash 2000 analyser (Thermo Fisher Scientific, Cambridge, UK). This method is based on combustion, where the carbon of the sample is converted into simple gas (CO2) in a pure oxygen atmosphere. The TC of the modified and activated CR samples was analysed with a Skalar Formacs HT TOC Analyzer (Skalar, Breda, Netherlands); TC was determined using catalytic oxidation at 1100 °C.
Sulphate ions were analysed in the filtrate solution by ion chromatography (Metrohm 761 Compact IC, Herisau, Switzerland), and some samples of preliminary and kinetic experiments were analysed using Vacu-vial tests (Sulfate Vacu-vials® Kit, CHEMetrics, Midland, VA, USA).
Zeta potential of the adsorbent suspension was measured with a Beckman Coulter Delsa™ Nano Submicron Particle Size and Zeta Potential Analyzer (Helsinki, Finland). A sample was placed in a 50-cm3 plastic vessel, into which 40 cm3 of a 10 mM NaCl solution was added. The contents were centrifuged for 10 min at 230 rpm. The pH values were adjusted using 0.1 M NaOH or HCl solutions. Surface functional groups were determined with a Perkin-Elmer Spectrum one FTIR (Turku, Finland).
Batch Sorption Experiments
The effects of initial pH, initial SO42- ion concentration, and contact time on the removal of SO42- ions were studied in each sorbent. Based on the pH sorption experiments, MCR 4 had the highest SO42- ion removal efficiency and was selected for subsequent experiments, including the effects of initial SO42-ion concentration and contact time. Unmodified CR and commercial AC were used as the reference samples. To determine the optimum initial pH for SO42- ion sorption by different sorbent materials, batch equilibrium studies were carried out at pH values of 2 to 8 using an initial concentration of 100 mg/L SO42- ion solution, which was prepared by diluting sodium sulphate (NaSO4·7H2O) in MilliQ water. The pH of the solution was adjusted with HCl and/or NaOH (FF Chemicals, Oulu, Finland). Because CR is a strongly alkaline material and possesses a high buffering capacity (Kilpimaa et al. 2013), pH adjustment was performed after adding the sorbent to the SO42- ion solution to ensure the correct pH during the sorption experiment. The bottles were placed on a laboratory shaker using a reciprocating motion at room temperature for 24 h. The optimum initial concentration of SO42- ion removal was selected by using different initial SO42- ion concentrations (50 to 1000 mg/L) at the optimum initial pH value. The pH and concentration optimisation experiments were performed using polyethylene flasks, and the sorbent dosage was 5.0 g/L.
When the pH and concentration optimisation experiments were completed, the effect of contact time was studied in the optimised conditions in a 2-L reactor vessel equipped with a magnetic stirrer at 1000 rpm, using a sorbent dosage of 5.0 g/L. Samples were collected after 1, 2, 5, 10, 20, and 30 min and 1, 2, 4, 6, 8, 12, and 24 h of contact time. All sorption experiments were performed at room temperature, and all pH and concentration optimisation experiments were duplicated. All samples, including the initial samples, were passed through 0.45-μm filter paper (Sartorius Stedim Biotech, Goettingen, Germany).
The percentage removal (%) of SO42- ions from the solution was calculated using Eq. 1,

where C0 and Ce are the initial and equilibrium concentrations, respectively, of SO42- in solution (mg/L). The sorption capacity qe (mg/g) after equilibrium was calculated using Eq. 2,
 (2)
(2)
where V is the volume of the solution (L) and m is the mass of the sorbent (g) (Meena et al. 2005; Bhatnagar et al. 2010a,b).
Adsorption Isotherms
Adsorption isotherms revealed the nature of the adsorption process at varying initial concentrations in optimal pH. The nonlinear Langmuir, nonlinear Freundlich, Sips, and bi-Langmuir models were applied to the experimental data. Isotherm parameters were obtained using a nonlinear regression model (GRG nonlinear function, Microsoft Excel software, Redmond, WA, USA). The Langmuir model assumes that monolayer adsorption takes place homogenously on equal sites where the adsorbates do not interact with each other. The general nonlinear form of the Langmuir equation is as follows,
 (3)
(3)
where qe is the equilibrium adsorption capacity (mg/g), qm is the maximum adsorption capacity of the adsorbent (mg/g), and bL is a constant related to the adsorption energy (L/mg) (Langmuir 1918; Repo et al. 2009).
The Freundlich isotherm is also used to describe adsorption from aqueous solutions. Freundlich expression is applicable to non-ideal adsorption on heterogeneous surfaces as well as multilayer adsorption. The Freundlich model is shown in Eq. 4,
 (4)
(4)
where Kf is a relative indicator of adsorption capacity (L/mg) and the dimensionless value 1/nf is the measure of surface heterogeneity becoming more heterogeneous as the value gets closer to zero (Freundlich 1906; Repo et al. 2009).
The Sips isotherm model is a combination of the Langmuir and Freundlich models. At low adsorbate concentrations, it reduces to the Freundlich isotherm, while in high coverages, it predicts monolayer adsorption in a similar manner as the Langmuir model. The Sips isotherm is given as follows,
 (5)
(5)
where bS is a constant related to the adsorption energy (L/mg) and ns is a dimensionless constant characterising the heterogeneity of the system (Sips 1948; Quintelas et al. 2009; Najafi et al. 2012; Salehi et al. 2012).
The bi-Langmuir isotherm was first suggested by Graham (1953) and has been applied in recent adsorption studies (Karthikeyan et al. 2004; Repo et al. 2009, 2011). While the bi-Langmuir isotherm is based on the theory of Langmuir, the model assumes that there are two favoured adsorption sites on the surface. Therefore, the expression (Eq. 6) is derived from the general form of the Langmuir isotherm:
 (6)
(6)
where qm1 and qm2 are the maximum adsorption capacities (mg/g) of two different adsorption sites. Similarly, bL1 and bL2 (mg/g) represent the energies of adsorption on these sites.
To evaluate the fit of the isotherm equations to the experimental data, different error functions were used: residual root mean square error (RMSE), sum of squares errors (SSE), and the chi-square test (χ2). A smaller error value indicated better curve fitting. The error functions were defined as follows (Han et al. 2009; Rostamian et al. 2011; Duman et al. 2015):
 (7)
(7)
 (8)
(8)
 (9)
(9)
where n is the number of experimental data, p is the number of parameters, and qe(exp) and qe(calc) are the experimental and calculated values, respectively, of adsorption capacity in equilibrium.
Kinetic Modeling
To analyse the sorption kinetics of SO42- ions in different sorbents, pseudo-first- and pseudo-second-order kinetic models were applied to the experimental data (Lagergren 1898; Ho and McKay 1999; Al Hamouz and Ali 2012). The first-order rate equation of Lagergren is used for liquid adsorption studies, as shown in Eq. 10,
 (10)
(10)
where qe and qt are the amounts of SO42- ion adsorbed (mg/g) at equilibrium (1440 min) and at time t(min), respectively, and kf is the pseudo-first-order rate constant (1/min). The linear form of the pseudo-second-order equation is expressed as follows:
 (11)
(11)
where ks is the pseudo-second-order rate equilibrium constant (g/mg min).
The effect of diffusion was studied using the intraparticle diffusion model based on Weber and Morris (1963). The model is represented as follows,
 (12)
(12)
where qt is the amount of SO42- ion on the surface of the sorbent (mg/g) at time t (min), ki is the intra-particle diffusion rate constant (mg g-1 min0.5), and C is the intercept (mg/g) (Crini et al. 2007; Bhatnagar et al. 2010b; Salehi et al. 2012).
RESULTS AND DISCUSSION
Effect of pH
The sulphate sorption capacity of sorbents treated in various ways was determined to evaluate the activation or modification methods. First, the effect of pH 2 to 8 was studied for all produced sorbents (ACR 1–3 and MCR 1–6) and reference samples (AC and CR) (Fig. 1). In all cases, the sorption capacity decreased as the solution became more neutral or basic. It is possible that the surface of the sorbent was highly protonated in an acidic medium, and therefore, tended to adsorb negative ions. In addition, OH– competes with SO42- ions for unoccupied surface sites at higher pH, which decreases the sorption of SO42- ions (Rui et al. 2011).
The SO42- ion removal efficiencies of the reference samples (AC and CR) are shown in Fig. 1a. For unmodified CR, the best removal efficiency (9%) was obtained at pH 2 using 100 mg/L SO42-. Based on this result, pH 2 was selected as the optimum pH value. Sulphate ion removal by AC was the highest under acidic conditions, but there were no significant differences in the removal capacity between pH 2 and 4. Furthermore, corresponding preliminary studies showed the same phenomenon, and therefore, a pH of 2 was selected as the optimum initial pH value for SO42- ion removal. The point of zero charge (pHzpc) for both sorbents, CR and AC, was 2.1. This pHzpc value describes the pH at which adsorbents have a net zero surface charge (Chang et al. 2010). This result suggests that acidic conditions are optimal for SO42- ion removal.
The SO42- ion removal efficiencies of activated and modified carbon residues (ACR 1-3 and MCR 1-6) are shown in Fig. 1b. Modified carbon residues (MCR 1-4) were better sorbents than activated carbon residues (ACR 1-3). Four sorbents (ACR 1-3 and MCR 5) did not remove any SO42- ions; their curves overlapped at 0% removal. Because MCR 4 showed the highest removal efficiency (82.2%) at an initial pH of 4, MCR 4 was selected for subsequent experiments. The pHzpc value of MCR 4 was 5.1, which indicated that the optimal condition for sulphate removal is under acidic conditions; therefore, a pH of 4 was selected as the optimum initial pH value.

Fig. 1. Total SO42- ion removal (%) vs. initial pH in a) reference samples, CR, and AC or b) different activated and modified carbon residues. The sorption studies were carried out at a constant temperature (room temperature) with a sorbent dose of 5 g/L, SO42- concentration of 100 mg/L, and contact time of 24 h.
Effect of Initial SO42- Ion Concentration
The different sorbents were tested for SO42- removal in an SO42- concentration range of 50 to 1000 mg/L and the optimum initial pH (Fig. 2). The removal efficiency was the highest at lower concentrations, indicating that specific adsorption sites were available for SO42- adsorption. At higher initial SO42- concentrations, the adsorbent was saturated. MCR 4 displayed the highest removal efficiency (~100%) at 50 mg/L SO42-. Likewise, CR and AC displayed 18.8 to 10.4% and 13.5 to 4.1% removal efficiencies, respectively, for in 50 to 1000 mg/L SO42-. The optimum initial SO42- ion concentrations for CR, MCR 4, and AC were 50, 50, and 200 mg/L, respectively.

Fig. 2. Effect of the initial SO42- ion concentration on adsorption. The optimum initial pH values were 4, 2, and 2 for MCR 4, CR, and AC, respectively. The sorption studies were carried out at a constant temperature (room temperature) with a sorbent dose of 5 g/L and contact time of 24 h.
Adsorption Isotherms
The nonlinear Langmuir, nonlinear Freundlich, Sips, and bi-Langmuir isotherm models were applied to the MCR 4 and AC experimental results (Fig. 3).
 Fig. 3. The sorption isotherms for a) MCR 4 and b) AC. The optimum initial pH for MCR 4 and AC was 4 and 2, respectively. The sorbent dose was 5.0 g/L, with 24-h contact at room temperature.
Fig. 3. The sorption isotherms for a) MCR 4 and b) AC. The optimum initial pH for MCR 4 and AC was 4 and 2, respectively. The sorbent dose was 5.0 g/L, with 24-h contact at room temperature.
The general isotherm models did not fit the unmodified CR experimental data, as its adsorption was relatively minor and varied with concentration. The Langmuir model poorly represented the MCR 4 data. This result was also seen in the low coefficient of determination value (R2) and high errors of the Langmuir curve (Table 1). According to the R2 values, the Freundlich and Sips isotherms provided a reasonable fit for the experiments, but the Sips model overestimated the qm value. Thus, the bi-Langmuir model was the most suitable model with an R2 value of 0.98. These results suggest the heterogeneous adsorption of SO42- anions onto the surface of MCR 4 with two different types of adsorption sites, and another of those adsorption sites have a slightly higher adsorption capacity and clearly higher affinities for the adsorption. These results support the conclusion made from the R2values of the Bi-Langmuir model (smallest R2) and the Langmuir model (largest R2). In the case of AC (Fig. 3b), the Sips model provided the best fit, with an R2 value of 0.95.
Table 1. Models of SO42- Ion Removal by Fe-Modified Carbon Residues (MCR 4) and Activated Carbon (AC)

Effect of Contact Time and Kinetic Modelling
The sorption of SO42- ions was studied as a function of time (1 min to 24 h) in the optimum initial pH and SO42- ion concentrations. The optimum initial pH values were 2, 4, and 2 for CR, MCR 4, and AC, respectively, and the optimum initial concentrations were 50, 50, and 200 mg/L for CR 4, MCR 4, and AC, respectively. The rate of SO42- ion removal was higher at the beginning of the sorption experiment because of a larger number of available adsorption sites (El-Ashtoukhy et al. 2008). Sulphate ion removal by MCR 4 was much greater than by AC. The highest removal efficiency (97.6%) and sorption capacity (10.3 mg/g) were achieved within about one minute with MCR 4; after this time, the removal efficiency decreased slightly. The CR kinetic data confirmed the adsorption isotherm analysis; however, CR did not work as an adsorbent because of its virtually non-existent adsorption efficiency.
Pseudo-first-order and pseudo-second-order models were applied to the data, the R2 value of the pseudo-first-order kinetic model was lower than that of the pseudo-second-order model. In addition, experimental uptake values (qe,exp) were not reasonable in regard to the calculated values (qe,calc). Therefore, the pseudo-second-order kinetic model was selected as the best-fit model, as shown in Table 2 and Fig. 4b.

Fig. 4. a) Effect of contact time on SO42- ion removal at the optimum initial pH and SO42- ion concentration. b) Pseudo-second-order kinetic plots. The sorbent dose was 5.0 g/L, and the sorption studies were carried out at room temperature. MCR 4: Fe-modified carbon residues; CR: carbon residues; AC: commercial activated carbon
Table 2. Summary of SO42- Removal and Pseudo-Second-Order Kinetics

MCR 4: Fe-modified carbon residues; CR: carbon residue; AC: commercial activated carbon; qe is the amount of SO42- ion adsorbed onto the sorbent at equilibrium (mg/g); k2 is the rate constant for pseudo-second-order kinetics; R2 is the linear coefficient of determination.
In all cases, the experimental qe,exp values corresponded to the calculated qe,calc values. Furthermore, the linear regression values (R2) were higher than 0.99 for MCR 4 and AC, indicating that the kinetics of sorption were accurately described by the pseudo-second-order model. In the case of SO42- ion removal by MCR 4, the calculated rate constant was negative and was not included in Table 4. The negative rate constant can be explained by the fact that maximal SO42- ion removal efficiency was achieved in one minute; after which, it varied because of the pH adjustment, then decreased slightly (Fig. 4a). For CR, the R2 value was the lowest compared to the other sorbents.
Mechanism of the Sorption
The Weber and Morris intraparticle diffusion model (Weber and Morris 1963) was applied to the kinetic data for MCR 4 and AC. The model indicated two-phase sorption on MCR 4; however, there were three lines for AC, showing that sorption on this surface was three-phased (Fig. 5). In both cases, the first stage was attributed to instantaneous or external surface sorption. In the case of AC, there were also phases of slower sorption, in which the adsorbates diffused into the inner pores of the sorbent material (Bhatnagar et al. 2010b). Finally, the sorption slowed down because of the low SO42-ion concentration or limitation of the sorbent.
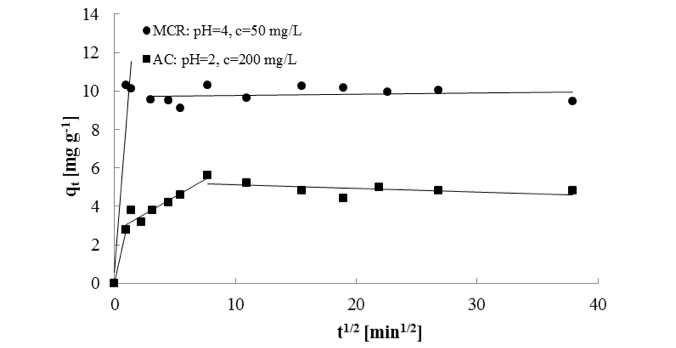
Fig. 5. Weber and Morris intraparticle diffusion model plots of SO42- ions on MCR 4 and AC
The adsorption mechanism is dependent on the pH and characteristics of the surface. For example, AC includes typically heteroatoms, such as oxygen, hydrogen, nitrogen, and sulphur, which influence the charge, hydrophobicity, and electronic density of the adsorbent surface. In the case of AC, the removal of ions from an aqueous solution is based on electrostatic adsorbate-adsorbent interactions, which are totally dependent on the carbon surface functionality. Other mechanisms enhance adsorption potential, which typically occurs in the narrowest microporosity and may be strong enough to adsorb ions (Dias et al. 2007). AC has a large surface area and large micropore volume (Table 3). However, FT-IR measurements suggested that AC had no functional groups. Thus, it was assumed that SO42- removal was primarily based on the porosity of the material.
For various industrial adsorbents, such as CR, the sorption mechanism can be very complicated. According to Ahmaruzzaman (2011), adsorption is attributable to electrostatic attraction, ion exchange, adsorption–precipitation, hydrogen bonding, and chemical interactions between impurities. Carbon residues from the biogasification process contain many components, such as calcium (42.3 g/kg), sodium (71 g/kg), and zinc (66.1 g/kg), which affect adsorption capacity (Kilpimaa et al. 2013); however, those components are dissolved during the acid washing step. The surface functional groups of various industrial adsorbents can also affect the sorption process. However, functional groups were not found by FT-IR of CR and MCR 4. Specific surface area, pore size, and pore volume are also important physical properties.
As shown in Table 3, CR and MCR 4 exhibited almost the same physical and chemical properties. They also exhibited much smaller surface areas and total pore volume (micro, meso, and macro) than AC. However, MCR 4 was a much better sorbent material for SO42- removal than CR and AC. Based on this finding, it was clear that impregnated iron plays a major role in the SO42- removal process.
According to Adhoum and Monser (2002), the removal mechanism becomes more complicated when adding impregnating chemicals to the sorbent material. In addition to adsorption, impurities can be removed by ion exchange, precipitation, and/or chemisorption on the newly-added chemicals.
Table 3. Physical and Chemical Properties of Adsorbents

MCR 4: Fe-modified carbon residues; CR: carbon residues; AC: commercial activated carbon
Comparison with Other Sorbents
When MCR 4 and AC was compared with other sorbents (Table 4), they exhibited comparable sorption capacity for SO42- ion removal.
Table 4. Comparison of Maximum Sorption Capacities for SO42- Ions

aLangmuir maximum sorption capacity, qm,calc
bExperimental maximum sorption capacity, qm,exp
The kinetic models of MCR 4 and AC were also compared with sorbents from other studies. According to Rui et al. (2011), Namasivayam and Sureshkumar (2007), and Namasivayam and Sangeetha (2008), the pseudo-second-order kinetic model fit the experimental data when surfactant-modified palygorskite, surfactant-modified coir pith, and ZnCl2-activated coconut coir pith were used as sorbents. The pseudo-second-order kinetic models were also used for the MCR 4 and AC data.
CONCLUSIONS
- Carbon residue wastes from a biomass gasification were chemically modified with iron chloride to remove SO42- ions from an aqueous solution, and results were compared with commercial activated carbon and unmodified carbon residue.
- Modified carbon residue is a potential sorbent for SO42- ion removal and it removed SO42- ions better than AC in the concentration range of 50 to 1000 mg/L. Unmodified CR did not work as a sorbent.
- The initial pH of 4 was optimal for MCR 4, and the maximum experimental sorption capacity (qexp) was 19.5 mg/g. For AC, the values were 2 and 7.59 mg/g, respectively.
- Sorption was modelled by the bi-Langmuir isotherm, and it follows the pseudo-second-order kinetic model.
ACKNOWLEDGMENTS
This study received financial support from the projects SULKA (A32164, 524/2012), HighBio2 (Biomass to fuels and chemicals/EU Interreg Nord, 304-8455-10), and Maa- ja vesitekniikan tuki ry. The authors are grateful to Mr. Mikko Niskanen for his contribution to some experiments. The authors also thank Mr. Jaakko Pulkkinen and Mr. Tuomo Vähätiitto for their assistance in SO42- ion and metal analysis and Dr. Pekka Tynjälä for his assistance in BET and TC measurements.
REFERENCES CITED
Adhoum, N., and Monser, L. (2002). “Removal of cyanide from aqueous solution using impregnated activated carbon,” Chem. Eng. Process.: Process Intensification 41(1), 17-21. DOI: 10.1016/S0255-2701(00)00156-2
Ahmadpour, A., and Do, D. D. (1996). “The preparation of active carbons from coal by chemical and physical activation,” Carbon 34(4), 471-479. DOI: 10.1016/0008-6223(95)00204-9
Ahmaruzzaman, M. (2011). “Industrial wastes as low-cost potential adsorbents for the treatment of wastewater laden with heavy metals,” Adv. Colloid Interface Sci. 166(1-2), 36-59. DOI: 10.1016/j.cis.2011.04.005
Al Hamouz, O. C. S., and Ali, S. A. (2012). “Removal of heavy metal ions using a novel cross-linked polyzwitterionic phosphonate,” Sep. Purif. Technol. 98, 94-101. DOI: 10.1016/j.seppur.2012.07.019
Bhatnagar, A., Kumar, E., and Sillanpää, M. (2010a). “Nitrate removal from water by nano-alumina: Characterization and sorption studies,” Chem. Eng. J. 163(3), 317-323. DOI: 10.1016/j.cej.2010.08.008
Bhatnagar, A., Minocha, A. K., and Sillanpää, M. (2010b). “Adsorptive removal of cobalt from aqueous solution by utilizing lemon peel as biosorbent,” Biochem. Eng. J. 48(2), 181-186. DOI: 10.1016/j.bej.2009.10.005
Cao, W., Dang, Z., Zhou, X., Yi, X., Wu, P., Zhu, N., and Lu, G. (2011). “Removal of sulphate from aqueous solution using modified rice straw: Preparation, characterization and adsorption performance,” Carbohydr. Polym. 85(3), 571-577. DOI: 10.1016/j.carbpol.2011.03.016
Chang, Q., Lin, W., and Ying, W. (2010). “Preparation of iron-impregnated granular activated carbon for arsenic removal from drinking water,” J. Hazard. Mater. 184(1-3), 515-522. DOI: 10.1016/j.jhazmat.2010.08.066
Chen, W., Parette, R., Zou, J., Cannon, F. S., and Dempsey, B. A. (2007). “Arsenic removal by iron-modified activated carbon,” Water Res. 41(9), 1851-1858. DOI: 10.1016/j.watres.2007.01.052
Crini, G., Peindy, H. N., Gimbert, F., and Robert, C. (2007). “Removal of C.I. Basic Green 4 (Malachite Green) from aqueous solutions by adsorption using cyclodextrin-based adsorbent: Kinetic and equilibrium studies,” Sep. Purif. Technol. 53(1), 97-110. DOI: 10.1016/j.seppur.2006.06.018
Dias, J. M., Alvim-Ferraz, M. C. M., Almeida, M. F., Rivera-Utrilla, J., and Sánchez-Polo, M. (2007). “Waste materials for activated carbon preparation and its use in aqueous-phase treatment: A review,” J. Environ. Manage. 85(4), 833-846. DOI: 10.1016/j.jenvman.2007.07.031
Duman, O., Tunç, S., and Polat, T. G. (2015). “Determination of adsorptive properties of expanded vermiculite for the removal of C. I. Basic Red 9 from aqueous solution: Kinetic, isotherm and thermodynamic studies,” Appl. Clay. Sci. 109-110, 22-32. DOI: 10.1016/j.clay.2015.03.003
El-Ashtoukhy, E.-.Z., Amin, N. K., and Abdelwahab, O. (2008). “Removal of lead (II) and copper (II) from aqueous solution using pomegranate peel as a new adsorbent,” Desalination 223(1-3), 162-173. DOI: 10.1016/j.desal.2007.01.206
European Commission (EC) Directive 2008/98/EC. (2008). “Directive 2008/98/EC of the European parliament and of the council of 19 November 2008 on waste and repealing certain directives (Waste Framework Directive),” European Union, Strasbourg, France.
Freundlich, H. M. F. (1906). “Over the adsorption in solution,” J. Phys. Chem. 57, 385-471.
Geelhoed, J. S., Hiemstra, T., and Van Riemsdijk, W. H. (1997). “Phosphate and sulfate adsorption on goethite: Single anion and competitive adsorption,” Geochimica et Cosmochimica 61(12), 2389-2396. DOI: 10.1016/S0016-7037(97)00096-3
Graham, D. (1953). “The characterization of physical adsorption systems. I. The equilibrium function and standard free energy of adsorption,” J. Phys. Chem. 57 (7), 665-669. DOI: 10.1021/j150508a014
Han, R., Zhang, J., Han, P., Wang, Y., Zhao, Z., and Tang, M. (2009). “Study of equilibrium, kinetic and thermodynamic parameters about methylene blue adsorption onto natural zeolite,” Chem. Eng. J. 145(3), 496-504. DOI: 0.1016/j.cej.2008.05.003
Hartinger, L. (1994). Handbook of Effluent Treatment and Recycling for the Metal Finishing Industry, (2nd Ed.), ASM International, Novelty, OH, USA.
Ho, Y. S., and McKay, G. (1999). “Pseudo-second order model for sorption processes,” Process Biochem. 34(5), 451-465. DOI: 10.1016/S0032-9592(98)00112-5
Karthikeyan, K. G., Mandla, A., Wang, T. D., and Kalbasi, M. (2004). “Solution chemistry effects on orthophosphate adsorption by cationized solid wood residues,” Environ. Sci. Technol. 38(3), 904-911. DOI: 10.1021/es034819z
Kilpimaa, S., Kuokkanen, T., and Lassi, U. (2013). “Characterization and utilization potential of wood ash from combustion process and carbon residue from gasification process,” BioResources 8(1), 1011-1027. DOI: 10.15376/biores.8.1.1011-1027
Kilpimaa, S., Runtti, H., Kangas, T., Lassi, U., and Kuokkanen, T. (2014). “Removal of phosphate and nitrate over a modified carbon residue from biomass gasification,” Chem. Eng. Res. Design 92(10), 1923-1933. DOI: 10.1016/j.cherd.2014.03.019
Knoef, H. M. F. (ed.) (2005). Handbook Biomass Gasification, BTG Biomass Technology Group BV, Enschede, Netherlands.
Lagergren, S. (1898). “About the theory of so-called adsorption of soluble substances,” K. Sven. Vetenskapsakad. Handl. 24(4), 1-39.
Langmuir, I. (1918). “The adsorption of gases on plane surfaces of glass, mica and platinum,” J. Am. Chem. Soc. 40(9), 1361-1403. DOI: 10.1021/ja02242a004
Madzivire, G., Petrik, L. F., Gitari, W. M., Ojumu, T. V., and Balfour, G. (2010). “Application of coal fly ash to circumneutral mine waters for the removal of sulphates as gypsum and ettringite,” Minerals Eng. 23(3), 252-257. DOI: 10.1016/j.mineng.2009.12.004
Meena, A. K., Mishra, G. K., Rai, P. K., Rajagopal, C., and Nagar, P. N. (2005). “Removal of heavy metal ions from aqueous solutions using carbon aerogel as an adsorbent,” J. Hazard. Mater. 122(1-2), 161-170. DOI: 10.1016/j.jhazmat.2005.03.024
Ministry of Social Affairs and Health (MSAH). (2000). Sosiaali- ja terveysministeriön asetus talousveden laatuvaatimuksista ja valvontatutkimuksista (in Finnish), (http://www.finlex.fi), Accessed April 2014.
Moret, A., and Rubio, J. (2003). “Sulphate and molybdate ions uptake by chitin-based shrimp shells,” Minerals Eng. 16(8), 715-722. DOI: 10.1016/S0892-6875(03)00169-9
Mulinari, D. R., and da Silva, M. L. C. P. (2008). ”Adsorption of sulphate ions by modification of sugarcane bagasse cellulose,” Carbohydr. Polym. 74(3), 617-320. DOI: 10.1016/j.carbpol.2008.04.014
Najafi, M., Yousefi, Y., and Rafati, A. A. (2012). “Synthesis, characterization and adsorption studies of several heavy metal ions on amino-functionalized silica nano hollow sphere and silica gel,” Seper. Purif. Technol. 85, 193-205. DOI: 10.1016/j.seppur.2011.10.011
Namasivayam, C., and Sureshkumar, M. V. (2007). “Removal of sulfate from water and wastewater by surfactant-modified coir pith, an agricultural solid “waste” by adsorption methodology,” J. Environ. Eng. Manage. 17(2), 129-135.
Namasivayam, C., and Sangeetha, D. (2008). “Application of coconut coir pith for the removal of sulfate and other anions from water,” Desalination 219(1-3), 1-13. DOI: 10.1016/j.desal.2007.03.008
Quintelas, C., Rocha, Z., Silva, B., Fonseca, B., Figueiredo, H., and Tavares, T. (2009). “Removal of Cd(II), Cr(VI), Fe(III) and Ni(II) from aqueous solutions by an E. coli biofilm supported on kaolin,” Chem. Eng. J. 149(1-3), 319-324. DOI: 10.1016/j.cej.2008.11.025
Rayner-Canham, G. (1999). Descriptive Inorganic Chemistry, 2nd Edition, W.H. Freeman, New York, NY, USA.
Repo, E., Petrus, R., Sillanpää, M., and Warchoł, J. K. (2011). “Equilibrium studies on the adsorption of Co(II) and Ni(II) by modified silica gels: One-component and binary systems,” Chem. Eng. J. 172(1), 376-385. DOI: 10.1016/j.cej.2011.06.019.
Repo, E., Kurniawan, T. A., Warchol, J. K., and Sillanpää, M. E. T. (2009). “Removal of Co(II) and Ni(II) ions from contaminated water using silica gel functionalized with EDTA and/or DTPA as chelating agents,” J. Hazard. Mater. 171(1-3), 1071-1080. DOI: 10.1016/j.jhazmat.2009.06.111
Rostamian, R., Najafi, M., and Rafati, A. A. (2011). “Synthesis and characterization of thiol-functionalized silica nano hollow sphere as a novel adsorbent for removal of poisonous heavy metal ions from water: Kinetics, isotherms and error analysis,” Chem. Eng. J. 171(3), 1004-1011. DOI: 10.1016/j.cej.2011.04.051
Rui, D., Yuanfa, L., Xingguo, W., and Jianhua, H. (2011). “Adsorption of sulfate ions from aqueous solution by surfactant-modified palygorskite,” J. Chem. Eng. Data 56(10), 3890-3896. DOI: 10.1021/je200544n
Salehi, E., Madaeni, S. S., and Heidary, F. (2012). “Dynamic adsorption of Ni(II) and Cd(II) ions from water using 8-hydroxyquinoline ligand immobilized PVDF membrane: Isotherms, thermodynamics, and kinetics,” Seper. Purif. Technol. 94, 1-8. DOI: 10.1016/j.seppur.2012.04.004
Sang, P., Wang, Y., Zhang, L., Chai, L., and Wang, H. (2013). “Effective adsorption of sulfate ions with poly(m-phenylenediamine) in aqueous solution and its adsorption mechanism,” Trans. Nonferrous Met. Soc. China 23(1), 243-252. DOI: 10.1016/S1003-6326(13)62452-8
Silva, A. M., Lima, R. M. F., and Leão, V. A. (2012). “Mine water treatment with limestone for sulfate removal,” J. Hazard. Mater. 221-222, 45-55. DOI: 10.1016/j.jhazmat.2012.03.066
Sips, R. (1948). “On the structure of a catalyst surface,” J. Chem. Physics 16(5), 490-495. DOI: 10.1063/1.1746922
Weber, Jr., W. J., and Morris, J. C. (1963). “Kinetics of adsorption on carbon from solution,” J. Sanit. Eng. Div. 89, 31-59.
Wu, C., Kuo, C., Lin, C., and Lo, S. (2002). “Modeling competitive adsorption of molybdate, sulfate, selenate, and selenite using a Freundlich-type multi-component isotherm,” Chemosphere 47(3), 283-292. DOI: 10.1016/S0045-6535(01)00217-X
Article submitted: November 27, 2015; Peer review completed: January 31, 2016; Revised version received and accepted: February 3, 2016; Published: February 9, 2016.
DOI: 10.15376/biores.11.2.3136-3152