
Abstract
To examine the effect of the presence of γ-hydroxymethyl groups on the rate of the β–O-4 bond cleavage during the alkaline pulping process, the rates of the β–O-4 bond cleavage of non-phenolic lignin model compounds without a γ-hydroxymethyl group, 2-(2-methoxyphenoxy)-1-(3,4-dimethoxyphenyl)ethanol (G’G) and 2-(2,6-dimethoxyphenoxy)-1-(3,4-dimethoxyphenyl)ethanol (G’S), were compared with those of analogous model compounds with a γ-hydroxymethyl group, 2-(2-methoxyphenoxy)-1-(3,4-dimethoxyphenyl)propane-1,3-diol (GG) and 2-(2,6-dimethoxyphenoxy)-1-(3,4-dimethoxyphenyl)propane-1,3-diol (GS), under alkaline pulping conditions. The disappearance of G’G or G’S was accompanied by the quantitative liberation of 2-methoxyphenol or 2,6-dimethoxyphenol, respectively, indicating that the disappearance resulted from the β–O-4 bond cleavage. The disappearance rate of G’G or G’S was in between those of the erythro and threo isomers of GG or GS, respectively. This result seems to be reasonably explained when the steric repulsions of the three staggered conformations are taken into consideration. The disappearance rate of G’G or G’S increased, but the increment became moderate with increasing hydroxide concentration.
Download PDF
Full Article
Quantitative Difference in the Rates of the β–O-4 Bond Cleavage between Lignin Model Compounds with and without γ-Hydroxymethyl Groups during the Alkaline Pulping Process
Satoko Shimizu, Pattaraporn Posoknistakul,a Tomoya Yokoyama,* and Yuji Matsumoto
To examine the effect of the presence of γ-hydroxymethyl groups on the rate of the β–O-4 bond cleavage during the alkaline pulping process, the rates of the β–O-4 bond cleavage of non-phenolic lignin model compounds without a γ-hydroxymethyl group, 2-(2-methoxyphenoxy)-1-(3,4-dimethoxyphenyl)ethanol (G’G) and 2-(2,6-dimethoxyphenoxy)-1-(3,4-dimethoxyphenyl)ethanol (G’S), were compared with those of analogous model compounds with a γ-hydroxymethyl group, 2-(2-methoxyphenoxy)-1-(3,4-dimethoxyphenyl)propane-1,3-diol (GG) and 2-(2,6-dimethoxyphenoxy)-1-(3,4-dimethoxyphenyl)propane-1,3-diol (GS), under alkaline pulping conditions. The disappearance of G’G or G’S was accompanied by the quantitative liberation of 2-methoxyphenol or 2,6-dimethoxyphenol, respectively, indicating that the disappearance resulted from the β–O-4 bond cleavage. The disappearance rate of G’G or G’S was in between those of the erythro and threo isomers of GG or GS, respectively. This result seems to be reasonably explained when the steric repulsions of the three staggered conformations are taken into consideration. The disappearance rate of G’G or G’S increased, but the increment became moderate with increasing hydroxide concentration.
Keywords: Alkyl-aryl ether; Cooking; Delignification; Hydrolysis
Contact information: Laboratory of Wood Chemistry, Department of Biomaterial Sciences, Graduate School of Agricultural and Life Sciences, The University of Tokyo, 1-1-1 Yayoi, Bunkyo-ku, Tokyo 113-8657, Japan; a: (Current address) School of Environment, Resources and Development, Asian Institute of Technology, P.O. Box 4, Klong Luang, Pathumthani 12120, Thailand;
*Corresponding author: yokoyama@woodchem.fp.a.u-tokyo.ac.jp
INTRODUCTION
It has been empirically demonstrated that hardwood is delignified more easily than softwood in an alkaline pulping process and that the efficiency of the process depends on the wood species, particularly for hardwoods. This fact suggests that a β–O-4 linkage (the alkyl-aryl ether linkage connecting the β-carbon of side chain and the aromatic #4 carbon, see Fig. 1) in lignin consisting of more syringyl units is cleaved more rapidly than one in lignin with fewer syringyl units, although many other factors can influence the delignification rate. It has been reported that there are wide variations in the proportions of the amounts of syringyl to guaiacyl aromatic nuclei (S/G ratio) and the erythro to threo stereo structures of the side-chains of β–O-4 type substructures (E/T ratio) present in hardwood lignin depending upon the wood species (Akiyama et al. 2005). Furthermore, a strongly positive correlation between the S/G and E/T ratios was determined from the structural analyses of lignin in various wood species (Akiyama et al. 2005). It has been suggested that the cleavage rate of a β–O-4 linkage of the erythro form is about 4 times as rapid as that of the threo counterpart, using not only model compounds (Miksche 1972; Tsutsumi et al. 1993; Criss et al. 2002) but also wood meal (Sugimoto et al. 2002). On the basis of this empirical and experimental information, it has become necessary to quantitatively understand the dependence of the chemical reactivity of β–O-4 substructures on the S/G and E/T ratios in an alkaline pulping process.
In our previous reports, lignin model compounds of C6-C3 type non-phenolic β–O-4 structures consisting of guaiacyl and syringyl aromatic nuclei were individually treated under alkaline pulping conditions (Shimizu et al. 2012). The model compounds used in this study were the erythro and threo isomers of 2-(2-methoxyphenoxy)-1-(3,4-dimethoxyphenyl)propane-1,3-diol (GG, Fig. 1), 2-(2,6-dimethoxyphenoxy)-1-(3,4-dimethoxyphenyl)propane-1,3-diol (GS, Fig. 1), 2-(2-methoxyphenoxy)-1-(3,4,5-trimethoxyphenyl)propane-1,3-diol (SG, Fig. 1), and 2-(2,6-dimethoxyphenoxy)-1-(3,4,5-trimethoxyphenyl)propane-1,3-diol (SS, Fig. 1). The results showed that the presence of a syringyl nucleus accelerates the β–O-4 bond cleavage; this accelerating effect is greater when the B-ring is a syringyl nucleus versus when the syringyl nucleus exists as the A-ring (see Fig. 1 for the definition of the A- and B-rings). The results also indicated that the β–O-4 bond of the erythro isomer is cleaved about 4 times more rapidly than that of the threo isomer for GG, while the ratio of the rate constant of the erythro to threo isomers ranges between 2 and 8 for the other model compounds, depending on the reaction temperature. Thus, it was confirmed that the cleavage rate is clearly dependent on the type of aromatic nucleus, i.e., syringyl or guaiacyl, and the stereostructure of the side-chain, i.e., erythro or threo and that these factors influence each other. It should be noted that the 4 times faster cleavage of β–O-4 bond of the erythro type (Miksche 1972; Tsutsumi et al. 1993; Criss et al. 2002) can be applied only to guaiacyl type lignin.

Fig. 1. Chemical structure of the model compounds used in this and our previous studies (Shimizu et al. 2012).
In this study, etherified phenolic lignin model compounds of the C6-C2 type containing a β–O-4 bond, 2-(2-methoxyphenoxy)-1-(3,4-dimethoxyphenyl)ethanol (G’G, Fig. 1) and 2-(2,6-dimethoxyphenoxy)-1-(3,4-dimethoxyphenyl)ethanol (G’S, Fig. 1), were individually treated under alkaline pulping conditions identical to those employed in our previous study (Shimizu et al. 2012). The side-chains of G’G and G’S lack the γ-hydroxymethyl group. Although similar etherified phenolic C6-C2 type lignin model compounds were used in previous studies to examine the β–O-4 bond cleavage under alkaline pulping conditions (Obst 1983; Hubbard et al. 1992; Collier et al. 1996; Criss et al. 1998; Schultz and Fisher 1998), the results of these studies cannot be compared with one another due to the different reaction conditions employed. The effect of the presence of the γ-hydroxymethyl group on the reaction rate is discussed by comparing the obtained results with those of our previous study (Shimizu et al. 2012), where GG and GS, which both have a γ-hydroxymethyl group, were used.
EXPERIMENTAL
Materials
All chemicals used were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan) and Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan). Water was deionized and degassed before use. The lignin model compounds (Fig. 1), G’G and G’S, were synthesized in accordance with the method of Adler et al. (1952). Two aromatic nuclei in each model compound are labeled as A- and B-rings (Fig. 1). The abbreviation of the model compounds is based on the type (guaiacyl (G) and syringyl (S)) of the A- and B-rings in this order. The 1H- and 13C-NMR spectra (JNM-A500, 500 MHz, JEOL Ltd., Tokyo, Japan) of these model compounds were recorded using acetone-d6 and aliquots of D2O as the solvents. The mass spectra of these model compounds were also analyzed using GC2010/PARVUM2 (GC/MS), Shimadzu Co., Kyoto, Japan. G’G: 1H-NMR δ 3.77, 3.80, 3.81 (s, 9H, -OCH3), 3.97 (dd, 1H, J = 8.0, J = 9.7, Cβ-Ha), 4.03 (dd, 1H, J = 3.8, J = 9.7, Cβ-Hb), 4.98 (dd, 1H, J = 3.8, J = 8.0, Cα-H), 6.83~7.00, 7.12 (m, 7H, aromatic); 13C-NMR δ 54.4, 54.5, 54.6 (-OCH3), 70.6 (Cα), 73.9 (Cβ), 109.6, 110.7, 111.5, 112.9, 117.8, 120.3, 120.5, 133.5, 147.6, 147.9, 148.3, 148.6 (aromatic); MS m/z (rel. int.) 304 (M+, 6), 286 (44), 271 (9), 257 (11), 226 (22), 211 (6), 180 (17), 167 (57), 151 (55), 139 (41), 124 (43), 109 (41), 91 (37), 77 (100), 65 (40), 51 (57). G’S: 1H-NMR δ 3.73 (dd, 1H, J = 9.0, J = 10.5, Cβ-Ha), 3.75, 3.77, 3.81 (s, 12H, -OCH3), 4.16 (dd, 1H, J = 3.5, J = 10.5, Cβ-Hb), 4.82 (dd, 1H, J = 3.5, J = 9.0, Cα-H), 6.66~6.68, 6.86~6.89, 7.00~7.02 (6H, aromatic); 13C-NMR δ 54.0, 54.1, 54.5 (-OCH3), 70.7 (Cα), 78.0 (Cβ), 104.3, 108.8, 110.3, 117.4, 122.8, 132.0, 135.7, 147.4, 147.8, 152.0 (aromatic); MS m/z (rel. int.) 334 (M+, 1), 316 (8), 287 (2), 272 (2), 256 (5), 180 (24), 167 (21), 154 (100), 151 (51), 139 (44), 124 (8), 121 (9), 117 (11), 93 (15), 91 (10), 77 (13), 65 (10).
Methods
A CH2Cl2 solution containing G’G or G’S (6.0 mmol/L) was prepared, and 1.0 mL of the solution was transferred to a stainless steel autoclave (10 mL volume, Taiatsu Techno® Co., Tokyo, Japan). The solution was dried under a current of N2; afterwards, 5.0 mL of a degassed NaOH solution was added to the autoclave. The concentration of the model compound was 1.2 mmol/L in the reaction solution. Then, the air in the autoclave was replaced with N2. After the autoclave was covered by a lid, it was immersed in an oil bath at 140 °C, 150 °C, 160 °C, or 170 °C, and the reaction was started. After the reaction was complete, the vessel was immediately immersed in ice water to cool the reaction solution. Then, acetic acid was added for neutralization.
Six vessels were prepared for conducting the reaction of each model compound at a prescribed temperature for six different reaction periods, and all the vessels were immersed together in the oil bath to perform the reactions together at the same temperature. All the reactions were repeated to confirm the reproducibility.
A CH3OH solution (2.0 mL) of an internal standard compound, 4-chlorophenol, was added to the autoclave containing the neutralized reaction solution. Because some solids appeared during the cooling process and did not disappear even after the addition of the CH3OH solution, the resulting mixture was extracted with 2.0 mL of CH2Cl2 three times in a glass tube. The combined organic layer was dried under a current of N2 in a round-bottomed flask, and 1.0 mL of CH3OH was added to dissolve the residue, followed by filtration.
The resulting mixture was analyzed by HPLC (LC-10A, Shimadzu Co., Kyoto, Japan) equipped with an SPD-M10A detector (280 nm, Shimadzu Co.) to quantify the residual lignin model compound and reaction product, 2-methoxyphenol (guaiacol) from G’G (Fig. 1) or 2,6-dimethoxyphenol (syringol) from G’S (Fig. 1), that was liberated from the B-ring of each model compound. The conditions for the HPLC analysis were as follows: column: Luna 5u C18(2) 100 A (150 mm x 4.6 mm, Phenomenex Inc., Torrance, CA, USA); oven temperature: 40 °C; flow rate: 1.0 mL/min; solvent system: gradient CH3OH/H2O (v/v) from 15/85 to 25/75 for 7.5 min, gradient to 54/46 for 37.5 min, gradient to 15/85 immediately and maintained for 5 min, total time: 50 min.
RESULTS AND DISCUSSION
General Description
In previous studies on alkaline delignification, organic co-solvents were used to completely dissolve the non-phenolic lignin model compounds. It has been suggested, however, that organic co-solvents affect the β–O-4 bond cleavage rate of these types of model compounds under alkaline cooking conditions (Obst 1983). In this study, no organic co-solvent was used, although G’G and G’S did not completely dissolve in the hydroxide solution when heated to around 80 °C. Because the disappearances of G’G and G’S in all the runs followed pseudo-first-order reactions after approximately 30 min of reaction (Fig. 2), it was reasonable to assume that the dissolution of G’G or G’S was complete during the latter half of the reaction and that the kinetics behaved as a homogeneous system during this time period.
The disappearances of G’G and G’S in the abovementioned periods of all the runs were approximated by pseudo-first-order kinetics, as representatively shown in Fig. 2. Table 1 lists the pseudo-first-order reaction rate constants (kobs), the Arrhenius activation energies (Ea), and the Arrhenius frequency factors (A) observed for G’G and G’S. Figure 3 illustrates the Arrhenius plots for G’G and G’S, which yielded the values of Ea and A. Because the reactions of G’G and G’S were conducted at several different temperatures, the rate constants (k) at 130 °C, 140 °C, 150 °C, 160 °C, and 170 °C were calculated for G’G and G’S based on the values of Ea and A (Table 2).
It is generally believed that a β–O-4 bond linking non-phenolic lignin units is cleaved via the neighboring group participation mechanism (Fig. 4) under alkaline pulping conditions. When the β–O-4 bond of G’G or G’S (also GG or GS) is cleaved, the reaction product, guaiacol or syringol, respectively, is liberated from the non-phenolic B-ring of each compound. Guaiacol and syringol were confirmed to be stable under the reaction conditions employed, when the oxygen in the autoclave as well as dissolved oxygen in the reaction solution was replaced with nitrogen before the reaction, which indicates that these compounds are not degraded by oxygen/alkaline-induced reactions (Shimizu et al. 2012). It was confirmed both in this study and our earlier study (Shimizu et al. 2012) that guaiacol or syringol is quantitatively detected accompanying the disappearance of G’G or G’S (or GG or GS in the Shimizu et al. 2012 study), respectively, when the air was replaced with nitrogen before the reaction. Therefore, the disappearance of G’G or G’S (also GG or GS) can quantitatively be attributed to the cleavage of the β–O-4 bond, most likely via the neighboring group participation mechanism.

Fig. 2. Representative logarithmic plot for the disappearance of G’G in the reaction where the concentration of NaOH was 1.0 mol/L and the temperature was 160 °C. [G’G]: concentration of G’G, [G’G]0: initial concentration of G’G

Fig. 3. Arrhenius plots of G’G and G’S in this study. ●: G’G, ○: G’S
Table 1. List of the Pseudo-First-Order Reaction Rate Constants (kobs), Arrhenius Activation Energy (Ea), and Arrhenius Frequency Factor (A) of G’G and G’S Observed in this Study

Table 2. List of the Rate Constants (k) of G’G and G’S Calculated Based on the Arrhenius Parameters

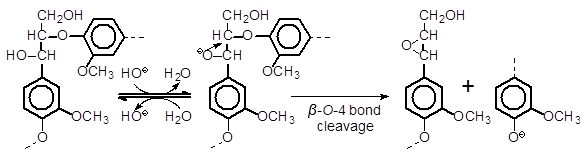
Fig. 4. The neighboring group participation mechanism for the β–O-4 bond cleavage of non-phenolic lignin substructures
Effect of the Presence of γ-Hydroxymethyl Groups on the Reaction Rate
In our previous study (Shimizu et al. 2012), the erythro and threo isomers of GG and GS (Fig. 1) were individually treated under alkaline pulping conditions identical to those employed in this study. GG and GS are constructed by replacing a β-hydrogen of G’G and G’S, respectively, with a hydroxymethyl group; this results in the group being in the γ-position relative to the A-ring. Table 3 lists the reaction rate constants (k), the Arrhenius activation energies (Ea), and the Arrhenius frequency factors (A) of GG and GS, which were determined in our previous investigation (Shimizu et al. 2012).
G’S always disappeared more rapidly than G’G, which indicates the accelerating effect of the presence of the syringyl nucleus as a B-ring (Table 2). This accelerating effect was also observed in our previous study using GS and GG (Shimizu et al. 2012). The ratio of the rate constant of G’S to that of G’G slightly decreased with increasing temperature, and was determined to be 1.9 and 1.8 at 130 °C and 170 °C, respectively. The ratios of the rate constant of GS to that of GG shown in our previous investigation (Shimizu et al. 2012) were 4.5 and 3.3 for the erythro and threo isomers, respectively, at 130 °C, and 2.6 and 2.4 for the erythro and threo isomers, respectively, at 170 °C. Thus, the effect of a syringyl nucleus for the B-ring on the rate of the β–O-4 bond cleavage was greater and more sensitive to temperature variation in the reactions of GG and GS than those of G’G and G’S. The increase in the steric repulsion caused by the substitution of a guaiacyl for a syringyl nucleus for the B-ring may be greater in the comparison between GG and GS carrying a γ-hydroxymethyl group than that between G’G and G’S without the γ-group. This can be a possible explanation for the observed phenomenon that the difference in the reaction rate between GG and GS was greater than that between G’G and G’S.
Table 3. List of the Rate Constants (k) of GG and GS Calculated Based on the Arrhenius Parameters in our Previous Report (Shimizu et al. 2012)

The disappearance rate of G’G and G’S was in between those of the erythro and threo isomers of GG and GS, respectively, except for the reaction of G’G at 130 °C. As shown in Fig. 4, the attack of the α-alkoxide on the β-carbon results in β–O-4 bond cleavage. This attack should be the rate-determining step. Therefore, the rate of the β–O-4 bond cleavage is affected by any factor that influences the rate of this attack. Several studies have indicated that the β–O-4 bond cleavage of the erythro isomer of lignin model compounds is faster than that of the corresponding threo isomers (Miksche 1972; Tsutsumi et al. 1993; Criss et al. 2002; Shimizu et al. 2012). The same phenomenon was also shown using wood meal (Sugimoto et al. 2002). This more rapid cleavage of the erythro isomer is commonly explained by the ease with which this isomer reaches the conformation where the relationship between the α-alkoxide and β-phenoxide is anti-periplanar; this favorable conformation aids the intramolecular SN2-type substitution reaction progress, as shown in line I of Fig. 5 (Criss et al. 1998). The steric repulsion of the conformation shown in line I for the threo isomer is larger than that in the erythro isomer.
There are three staggered conformations during the rotation about the carbon-carbon bond between the α– and β-carbon in erythro-GG, threo-GG, and G’G (Fig. 5). When the steric repulsions of these three conformations for the erythro-GG are considered, the conformation shown in line I, where the α-alkoxide can attack the β-carbon, is sterically the least crowded; the probability of reaching this conformation should be more than 1/3 in the erythro-GG isomer, when the probability of reaching conformations other than the staggered is assumed to be negligible. On the other hand, the probability of reaching the conformation shown in line I is the lowest for the threo-GG and should be less than 1/3. The conformation shown in line II seems to be the sterically most crowded among the three staggered conformations of G’G, although its steric repulsion may not be very different from those of the other two staggered conformations. Hence, the probability of reaching the conformation shown in line I for G’G may be more than 1/3. The steric repulsion of the conformation of G’G shown in line III does not seem to be different from that shown in line I. Therefore, the probability of reaching the conformation shown in line I for G’G may be more than 1/3, but less than that for erythro-GG. Judging from these considerations, it may seem reasonable that the rate of the β–O-4 bond cleavage of G’G was in between those of erythro– and threo-GG.

Fig. 5. Display of three staggered conformations of erythro-GG, threo-GG, and G’G during rotation about the carbon-carbon bonds between the α– and β-positions
Effect of Hydroxide Concentration on the Reaction Rates of G’G and G’S
The proportion of the amount of the α-alkoxide versus the undissociated α-alcohol, which is affected by the hydroxide concentration, is the other rate-determining factor in the neighboring group participation mechanism, although the γ-alkoxide may slightly contribute to this type of attack for lignin models containing a γ-hydroxylmethyl group (Taneda et al. 1987; Criss et al. 2002). However, it is not clear how the presence of a γ-hydroxylmethyl group and the erythro and threo diastereomers affects the acidity of the α-hydroxyl group. The contribution of the γ-alkoxide to the β–O-4 bond cleavage has been suggested to be relatively large in the reaction of the erythro isomer of a C6-C3 (phenylpropane) type model compound (Criss et al. 2002).
As described above, the β–O-4 bond cleavage should be accelerated by increasing the proportion of the α-alkoxide to that of the undissociated α-alcohol accompanying the increase of hydroxide concentration. Therefore, the dependence of the β–O-4 bond cleavage rate of G’G and G’S on hydroxide concentration was examined. The results are shown in Table 4. The disappearances of G’G and G’S were accelerated with increasing hydroxide concentration, although the increment of the reaction rate became moderate with the increase of hydroxide concentration. This was expected because the α-hydroxyl group should dissociate slightly in the relatively low hydroxide concentration and the dissociation should be enhanced with increasing hydroxide concentration. However, further dissociation should not be very sensitive to the increase in the relatively high hydroxide concentration, where the extent of the dissociation should already be relatively great. It is suggested that the variation of the hydroxide concentration employed in this study is within the range where the α-hydroxyl groups dissociate, although the pKa values of the α-hydroxyl groups are not exactly known. The ratio of the rate constants of G’S to that of G’G decreases with increasing hydroxide concentration, and is 2.1, 1.9, and 1.3 when the hydroxide concentration was 0.5, 1.0, and 2.0 mol/L, respectively. The difference in the disappearance rates between G’G and G’S decreases with increasing hydroxide concentration.
Table 4. List of the Pseudo-First-Order Reaction Rate Constants (kobs) of G’G and G’S Observed when the Hydroxide Concentration was Varied

The contribution of the alkali to the reaction rate can be evaluated using the reaction order n with respect to the hydroxide concentration in Equation (i), which can be arranged to Equation (ii). The reaction order n is expressed as the slope of a straight line obtained when ln[HOˉ] and ln(kobs) are assigned to the x- and y-axes, respectively, as shown in Fig. 6. The y-intercept is ln(kobs’). When the reaction order n is close to unity, the reaction rate is almost proportional to the hydroxide concentration. On the other hand, the reaction rate is nearly independent of hydroxide concentration when n is close to 0.
–d[model]/dt = kobs[model] = kobs’[HOˉ]n[model] (i)
[model]: concentration of G’G or G’S
[HOˉ]: concentration of alkali
kobs: pseudo-first-order reaction rate constant at each alkaline concentration
kobs’: rate constant (kobs’ = kobs/[HOˉ]n)
n: reaction order with respect to HOˉ
ln(kobs) = n ln[HOˉ] + ln(kobs’) (ii)
Table 5. List of the Reaction Orders (n), Rate Constants (kobs’), and Squares of the Correlation Coefficient (R 2) of G’G and G’S Calculated from Equation (ii)

* The values in the parentheses are those obtained when the highest hydroxide concentration for each compound was excluded from the approximation.
Table 5 lists the reaction orders n and rate constants kobs’ for G’G and G’S. The values in the parentheses are those obtained when the highest hydroxide concentration for each compound was excluded from the approximation. The approximation to a straight line was rather good when the highest hydroxide concentration was excluded, as can be seen from the values of R2and the dotted lines in Fig. 6. The reaction order n for G’G was close to unity, which indicated that the reaction rate was almost proportional to the alkaline concentration. On the other hand, the reaction order n for G’S was smaller than unity. The contribution of hydroxide concentration to the reaction rate was greater for G’G than for G’S. These results suggested that the proportion of ionized G’G α-alkoxide to G’G α-alcohol is more sensitive than the G’S model for the temperature and hydroxide concentration ranges examined. It is likely that the pKavalue of the α-hydroxyl group of G’G is slightly higher than that of G’S.

Fig. 6. Logarithmic plots for the correlation between the pseudo-first-order reaction rate constants and hydroxide concentration. ●: G’G, ○: G’S. Solid lines: approximation using 4 data points of each compound. Dotted lines: approximation using 3 data points, excluding the point at the highest hydroxide concentration
CONCLUSIONS
- 1. The difference in the disappearance rates between G’G and G’S was smaller than that between GG and GS, suggesting that the presence of a γ-hydroxymethyl group significantly increases the steric repulsion accompanying the substitution of a guaiacyl for a syringyl nucleus as the B-ring.
- The reaction rates of G’G and G’S were in between those of the erythro and threo isomers of GG and GS, respectively, which seems to be reasonable when the steric repulsions of the staggered conformations are taken into consideration.
- The reaction rates of G’G and G’S increased with increasing the alkaline concentration, but the increment became moderate with the increase of the alkaline concentration.
REFERENCES CITED
Adler, E., Lindgren, B. O., and Saeden, U. (1952) “The β-guaiacyl ether of α-veratrylglycerol as a lignin model,” Svensk Papperstidn. 55(7), 245-254.
Akiyama, T., Goto, H., Nawawi, D. S., Syafii, W., Matsumoto, Y., and Meshitsuka, G. (2005). “Erythro/threo ratio of β–O-4 structures as an important structural characteristic of lignin part 4: Variation in the erythro/threo ratio in softwood and hardwood lignins and its relation to syringyl/guaiacyl ratio,” Holzforschung 59(3), 276-281.
Collier, W. E., Fisher, T. H., Ingram, L. L., Harris, A. L., and Schultz, T. P. (1996). “Alkaline hydrolysis of nonphenolic β–O-4 lignin model dimers: Further studies of the substituent effect on the leaving phenoxide,” Holzforschung 50(5), 420-424.
Criss, D. L., Fisher, T. H., and Schultz, T. P. (1998). “Alkaline hydrolysis of nonphenolic α-carbonyl β–O-4 lignin dimmers substituted on the leaving phenoxide ring: Comparison with benzylic hydroxyl analogues,” Holzforschung 52(1), 57-60.
Criss, D. L., Elder, T., Fisher, T. H., and Schultz, T. P. (2002). “Effect of the α-and γ-hydroxyls on the alkaline hydrolysis rate of nonphenolic β–O-4 lignin diastereomers,” Holzforschung56(1), 67-72.
Hubbard, T. F., Schultz, T. P., and Fisher, T. H. (1992). “Alkaline hydrolysis of nonphenolic β–O-4 lignin model dimers: Substituent effects on the leaving phenoxide in neighboring group vs. direct nucleophilic-attack,” Holzforschung 46(4), 315-320.
Miksche, G. E. (1972). “Zum alkalischen abbau der p-alkoxy-arylglycerin-β-arylätherstrukturen des lignins: Versuche mit erythro-veratrylglycerin-β-guajacyläther,” Acta Chem. Scand.26(8), 3275-3281.
Obst, J. R. (1983). “Kinetics of alkaline cleavage of β-aryl ether bonds in lignin models: Significance to delignification,” Holzforschung 37(1), 23-28.
Schultz, T. P. and Fisher, T. H. (1998). “Phenoxyl substituent effect on the alkaline hydrolysis rates of β–O-4 lignin models: A review,” J. Pulp Pap. Sci. 24(8), 242-246.
Shimizu, S., Yokoyama, T., Akiyama, T., and Matsumoto, Y. (2012). “Reactivity of lignin with different composition of aromatic syringyl/guaiacyl structures and erythro/threo side chain structures in β–O-4 type during alkaline delignification: As a basis for the different degradability of hardwood and softwood lignin,” J. Agric. Food Chem. 60(26), 6471-6476.
Sugimoto, T., Akiyama, T., Matsumoto, Y., and Meshitsuka, G. (2002). “The erythro/threo ratio of β–O-4 structures as an important structural characteristic of lignin part 2: Changes in erythro/threo (E/T) ratio of β–O-4 structures during delignification reactions,” Holzforschung 56(4), 416-421.
Taneda, H., Nakano, J., Hosoya, S., Chang, H. (1987). “Stability of α-ether type model compounds during chemical pulping processes,” J. Wood Chem. Technol. 7(4), 485-497.
Tsutsumi, Y., Kondo, R., and Imamura, H. (1993). “Reaction of syringylglycerol-β-syringyl ether type of lignin model compounds in alkaline-medium,” J. Wood Chem. Technol. 13(1), 25-42.
Article submitted: May 10, 2013; Peer review completed: June 26, 2013; Revised version accepted: July 8, 2013; Published: July 9, 2013.